- 地球上约有 870 万种生物——形态各异,却共享同一套"语言":DNA
- 从微小的病毒,到体型庞大的北极熊
- 从路旁的蒲公英,到深海中的海龟
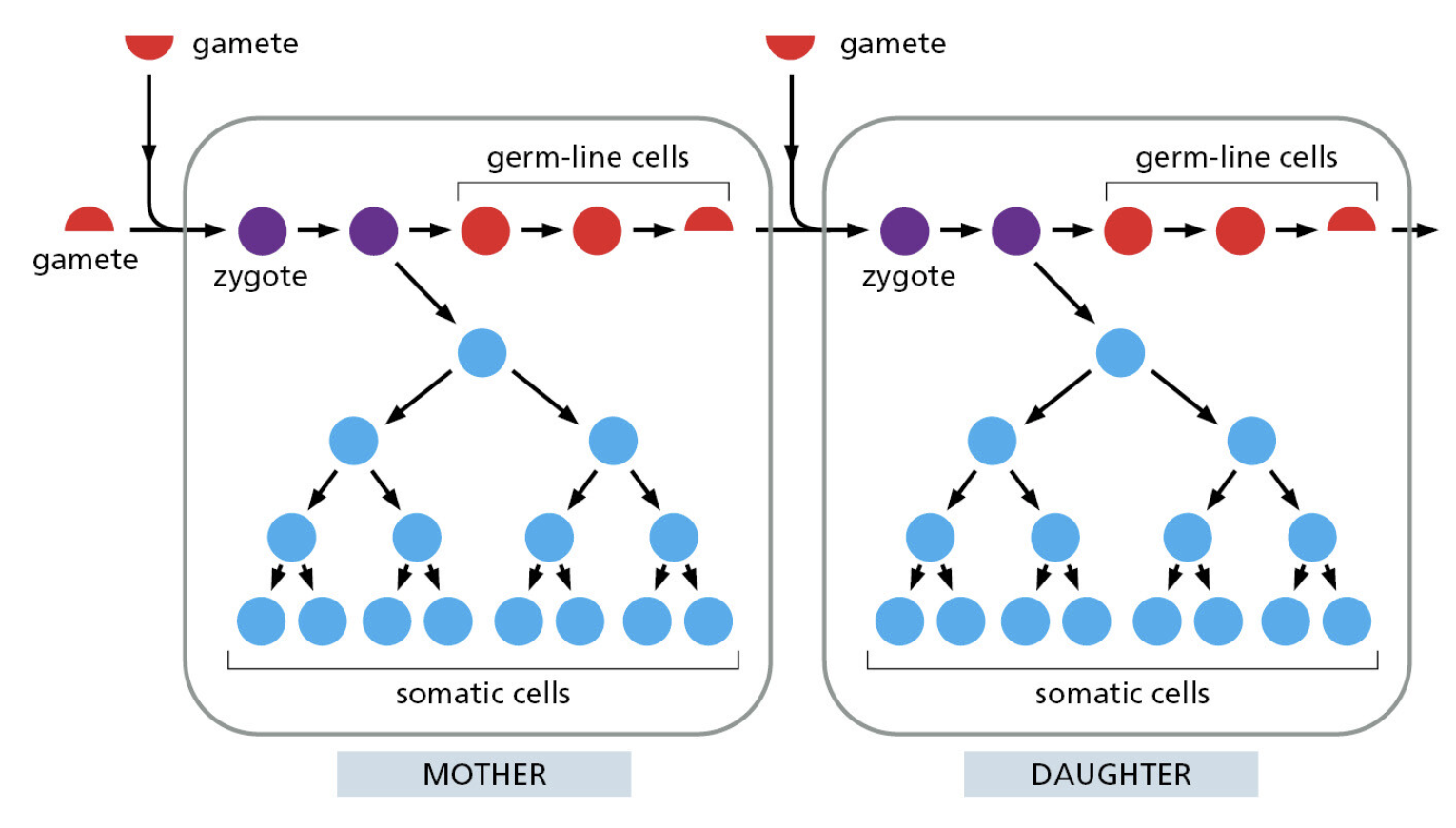
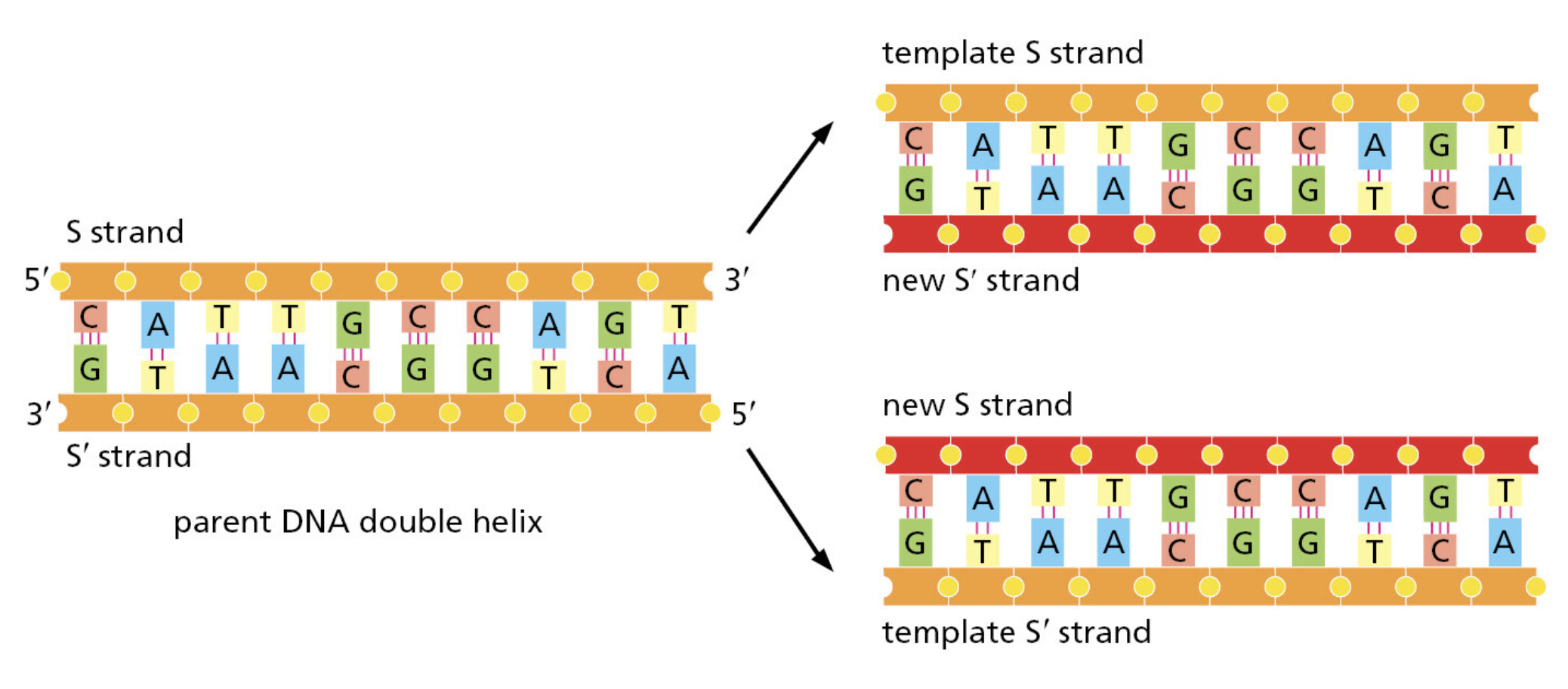
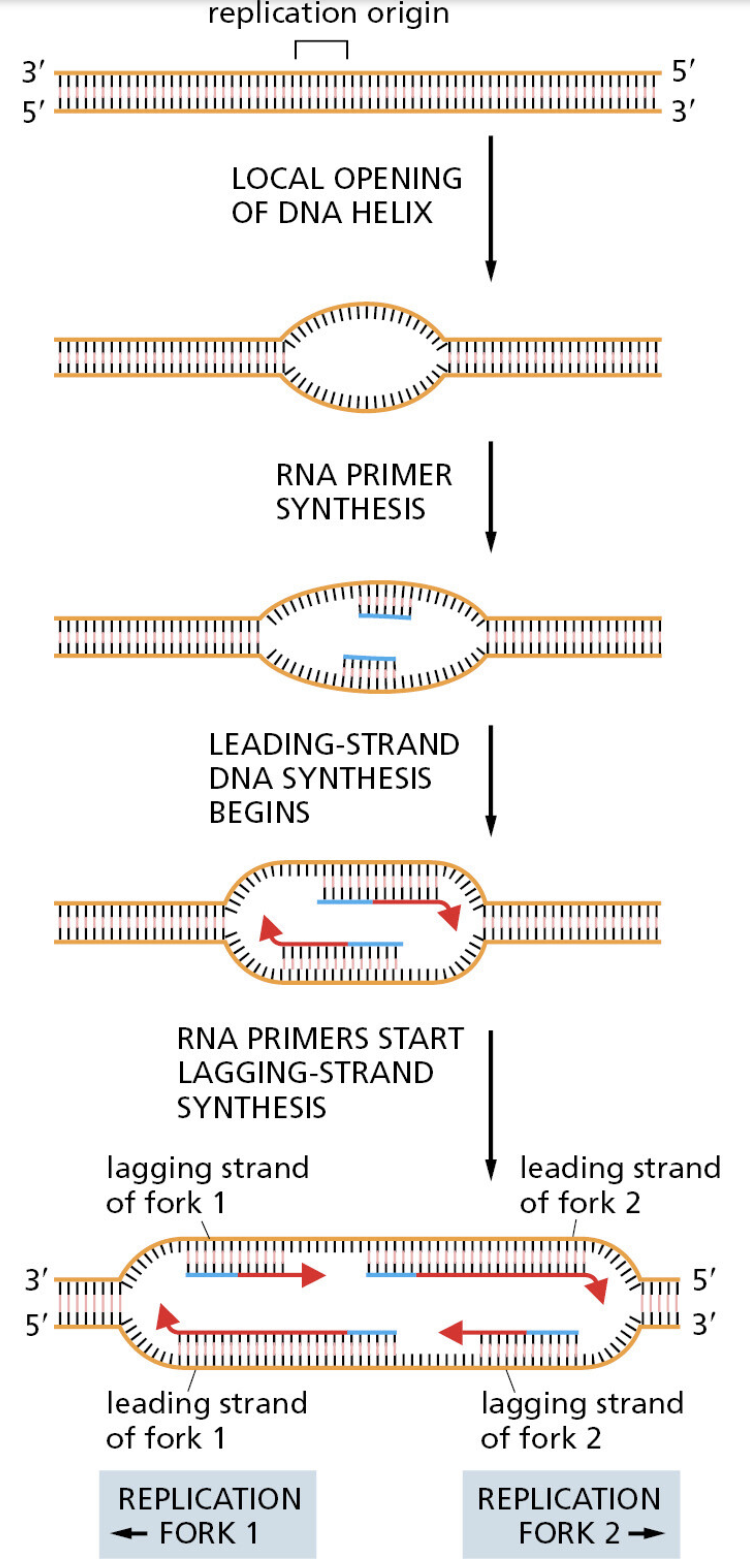
- 它们全都面对同一挑战:每次细胞分裂,都要将整个基因组精确复制一遍
- 你的身体每天复制"30亿个字母"——出错率哪怕只有 1%,就会产生约 3000 万处突变
- 为什么我们没有每天"变成另一种生物"?
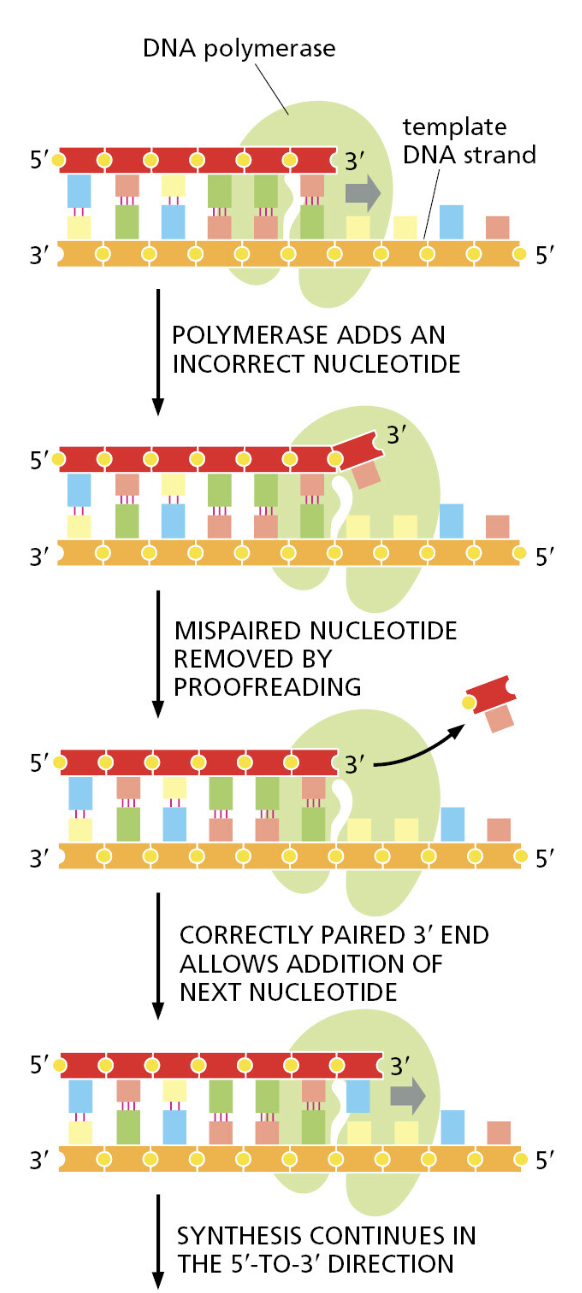
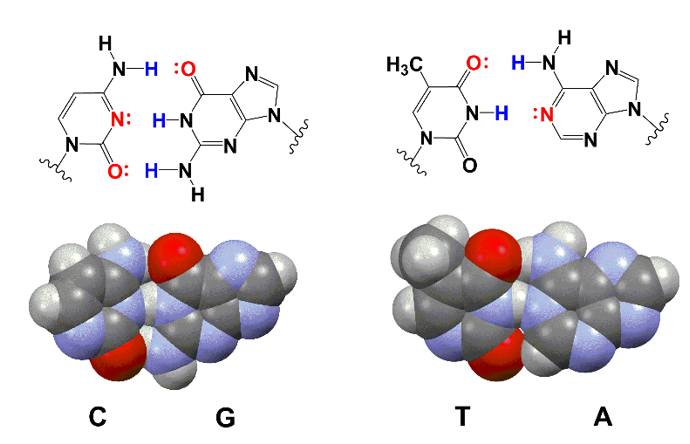
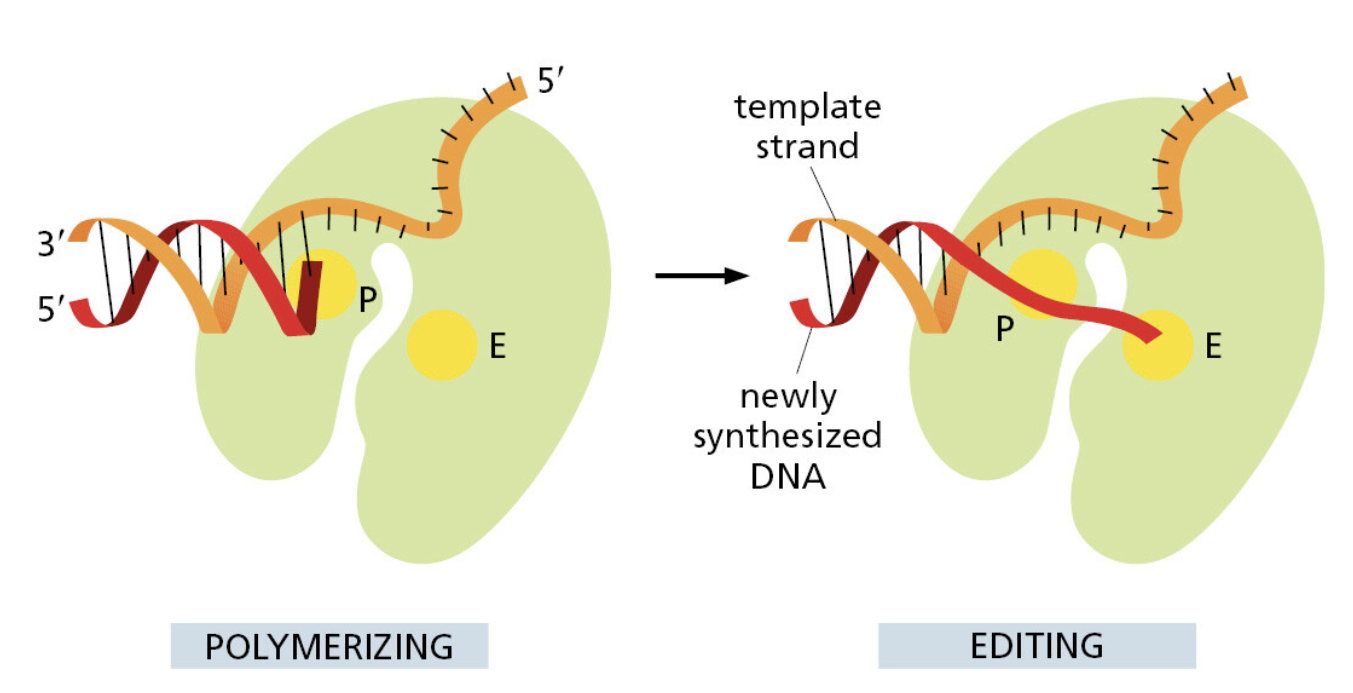
生命系统的第一原则:信息复制必须极其精准。

生命多样性
生命系统的第一原则:信息复制必须极其精准。
自然系统:经过数十亿年进化,DNA 复制已达到极致保真。
人类技术:我们刚刚开始学会在这套系统上"动手"。
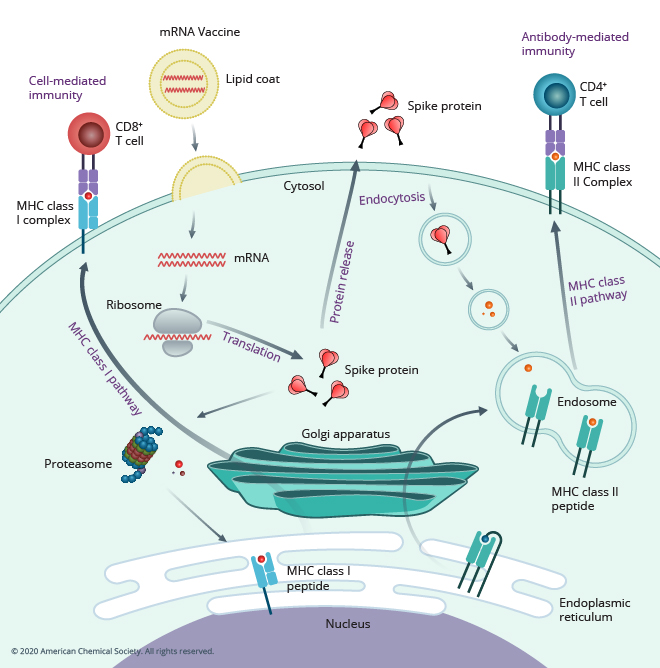
类比:疫苗不是替你"打怪",而是先教会免疫系统如何识别目标。
| 生物系统 | 类比 | 功能含义 |
|---|---|---|
| DNA | 硬盘 | 长期存储遗传信息 |
| RNA | 缓存 / U盘 | 临时调用与传递信息 |
| 蛋白质 | 程序执行体 | 真正完成细胞功能 |
关键点:DNA 本身不直接"干活",它提供的是说明书。
为什么需要 RNA:DNA 存于细胞核内需长期稳定保存,RNA 作为中间层让信息调用更灵活、更可调控。
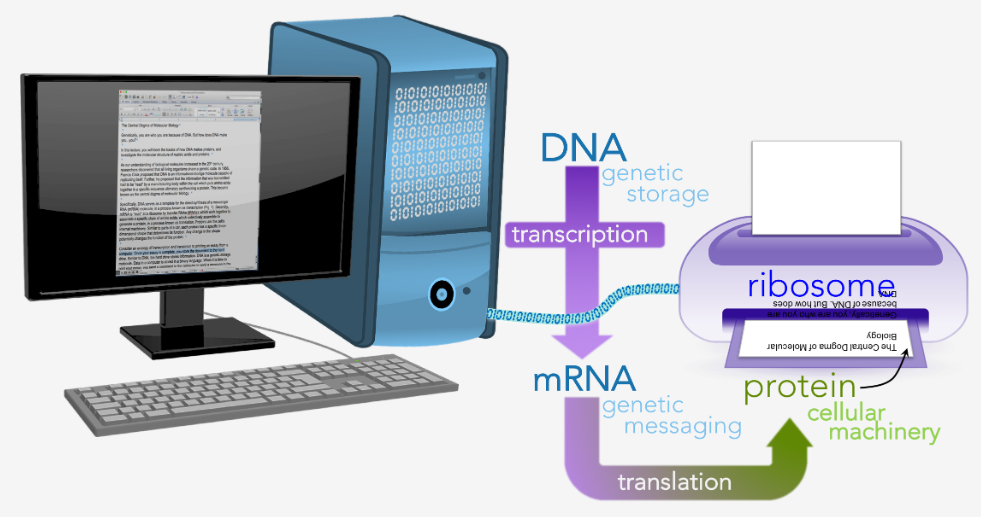
正常调控:细胞选择性读取DNA,精确的开关决定细胞身份。
治疗逻辑:靶向药瞄准异常蛋白,RNA疗法直接干预异常转录本。
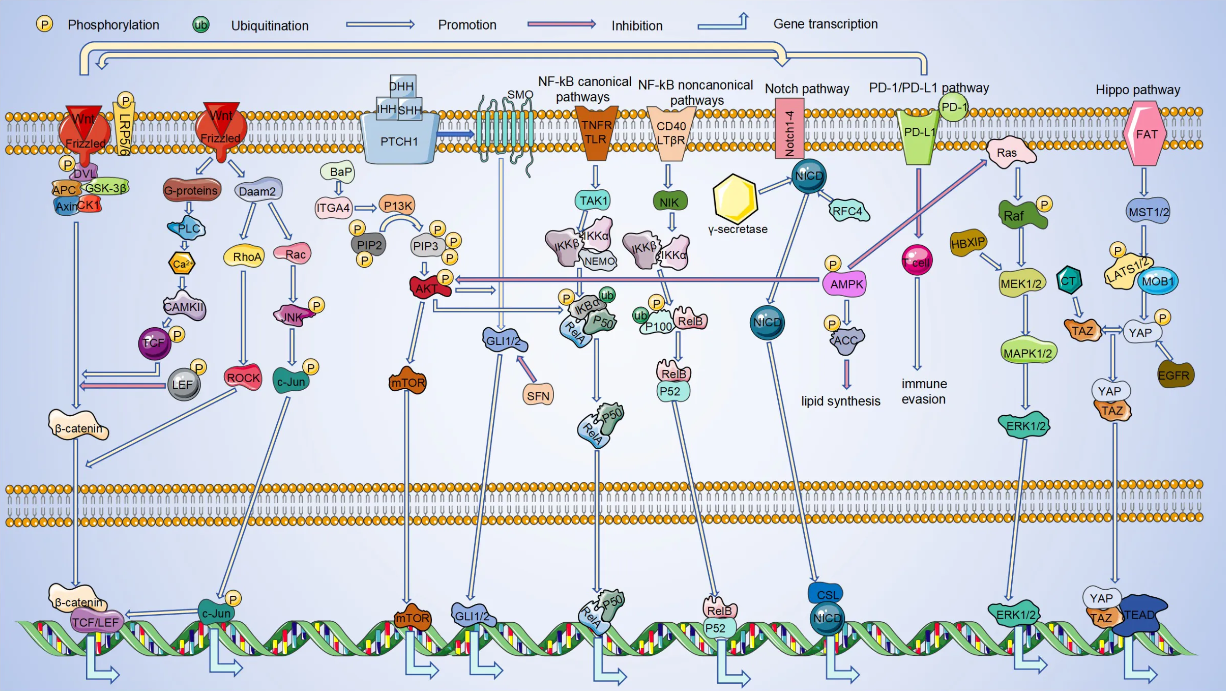
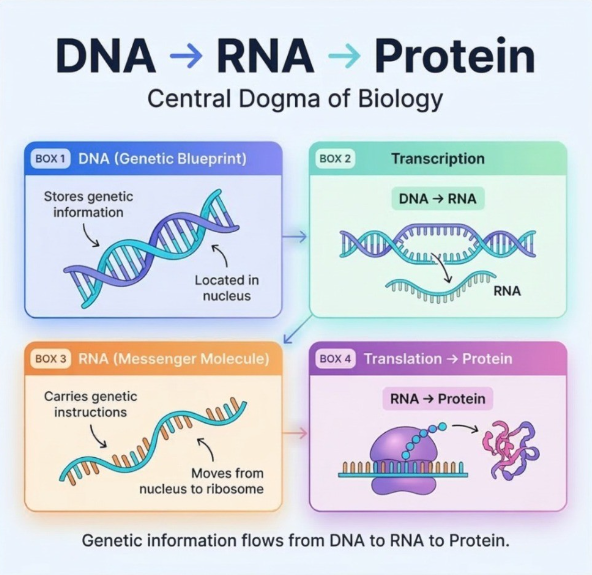
AI是理解生命语言的新工具。但真正的生物学问题仍是:DNA如何一步步变成蛋白质?
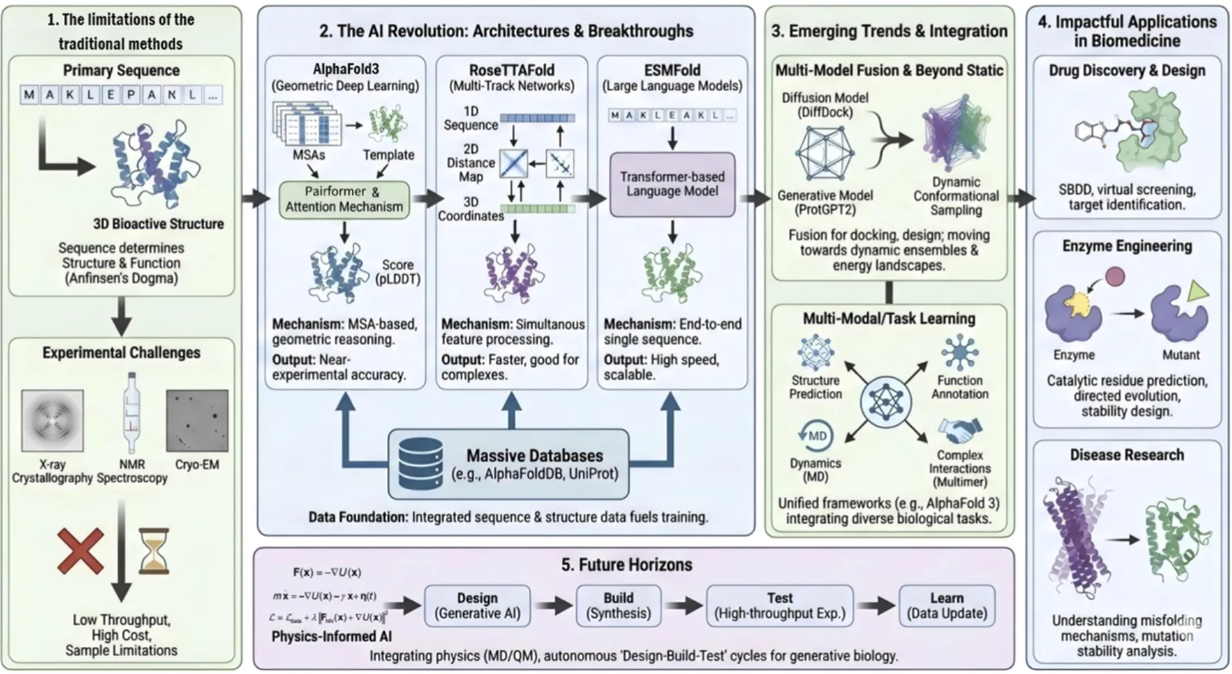
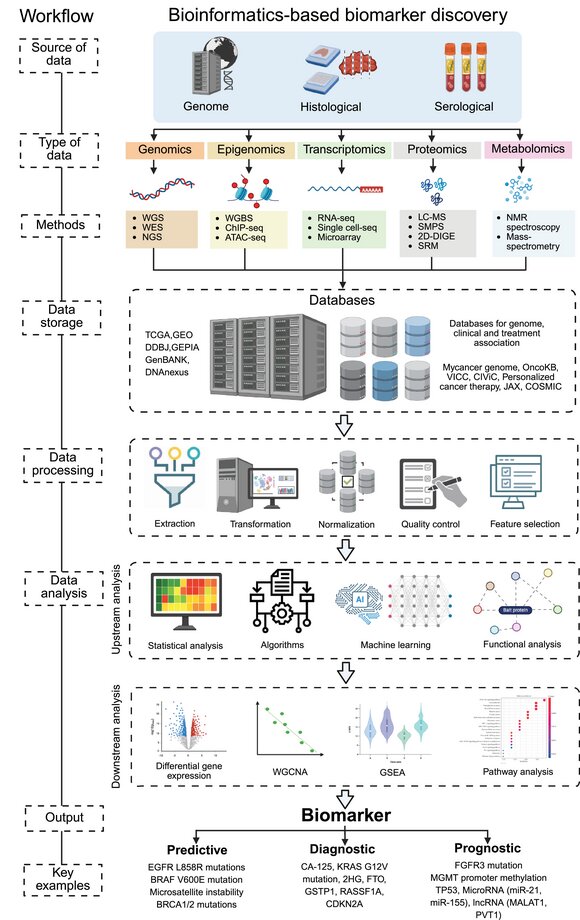
从基础研究到临床新药,核酸科学正在重塑现代医学。
这一切的底层逻辑,都指向同一个问题:核酸如何携带、传递并执行生命信息?

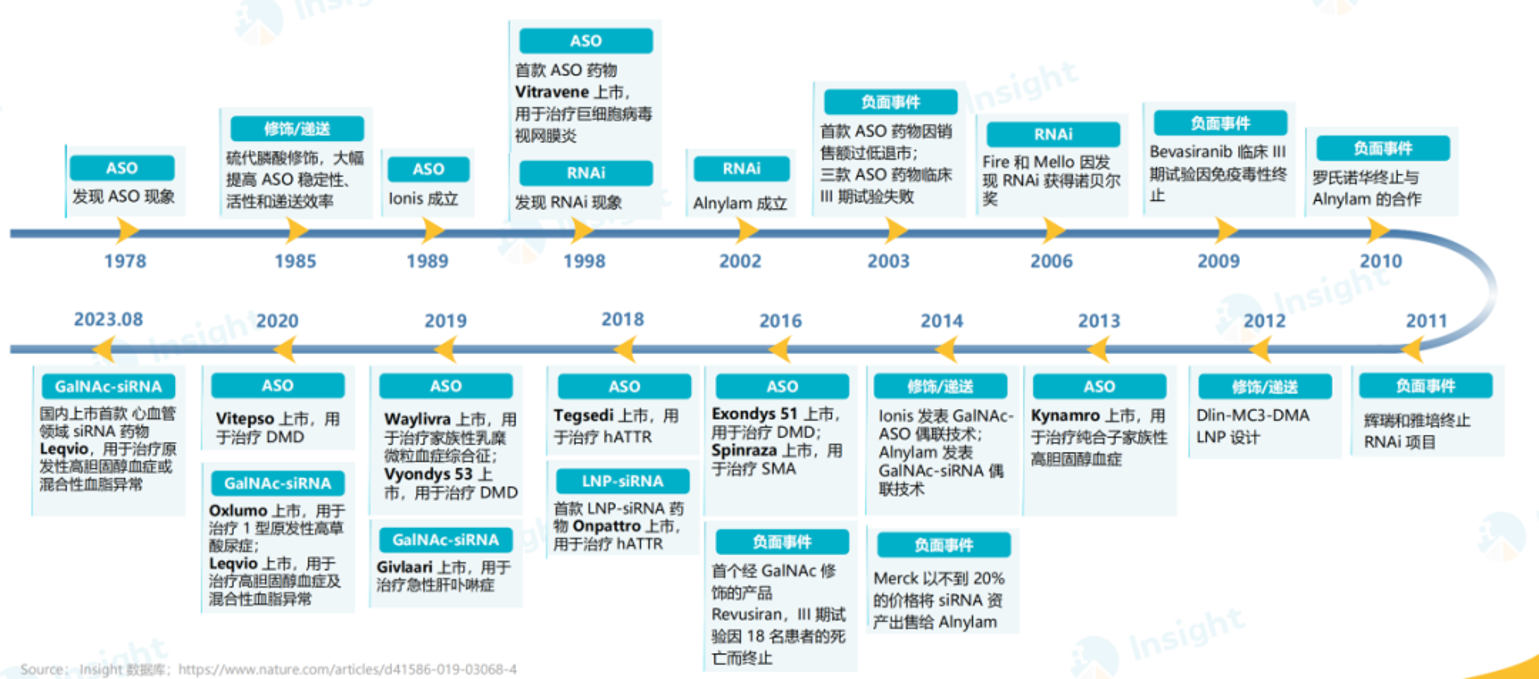
核酸药物产业正从"概念验证"走向"规模化落地",中国企业加速追赶国际前沿。
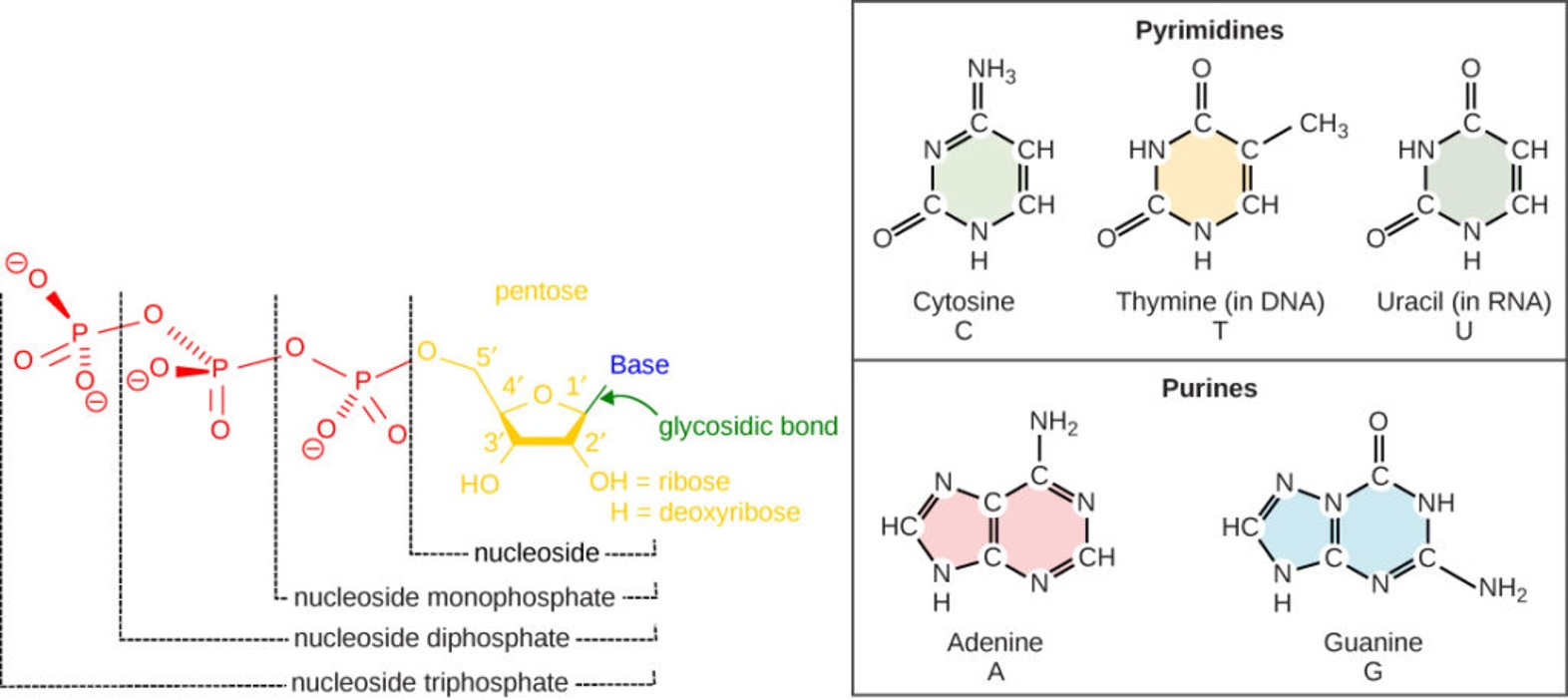
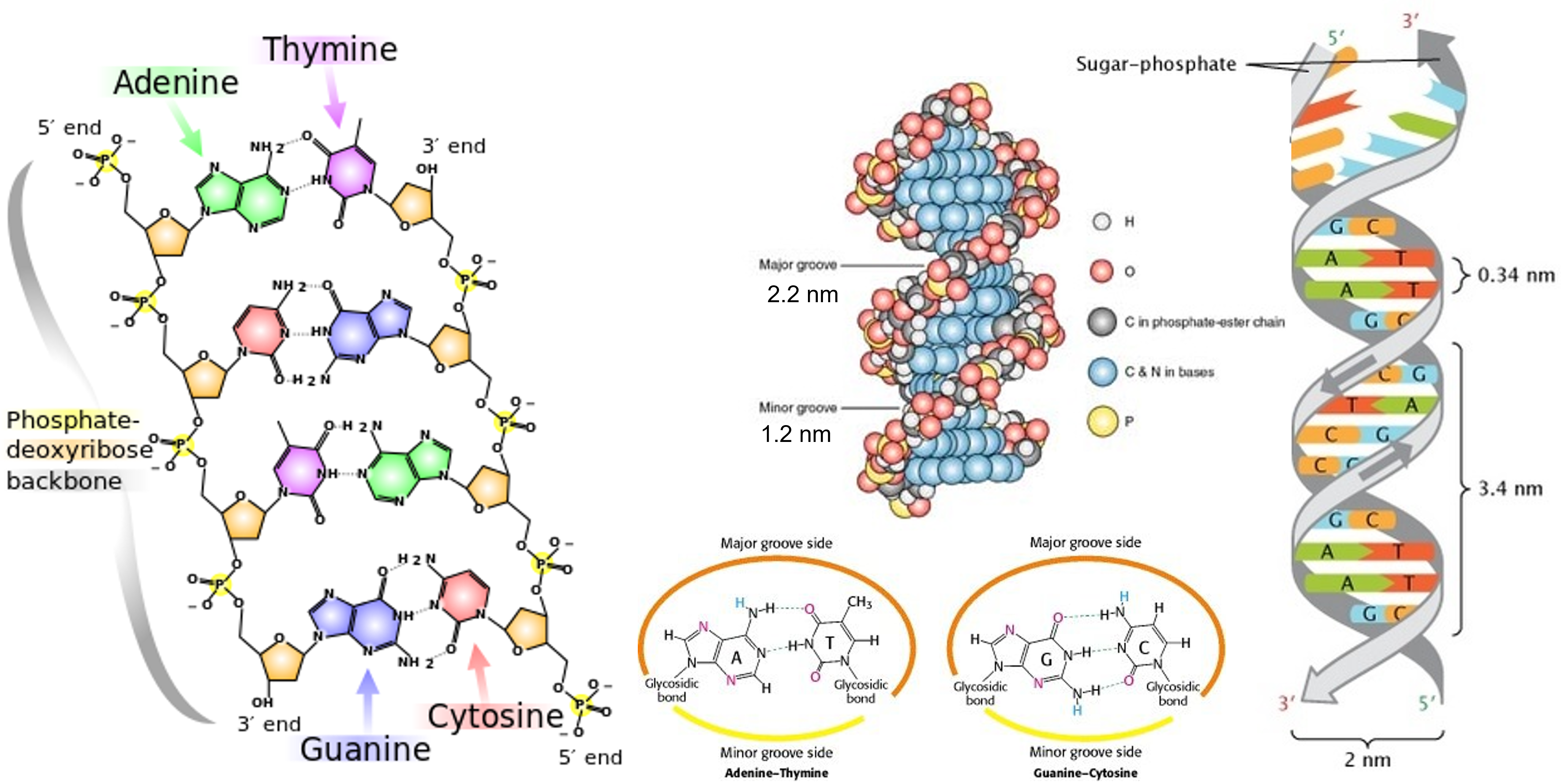

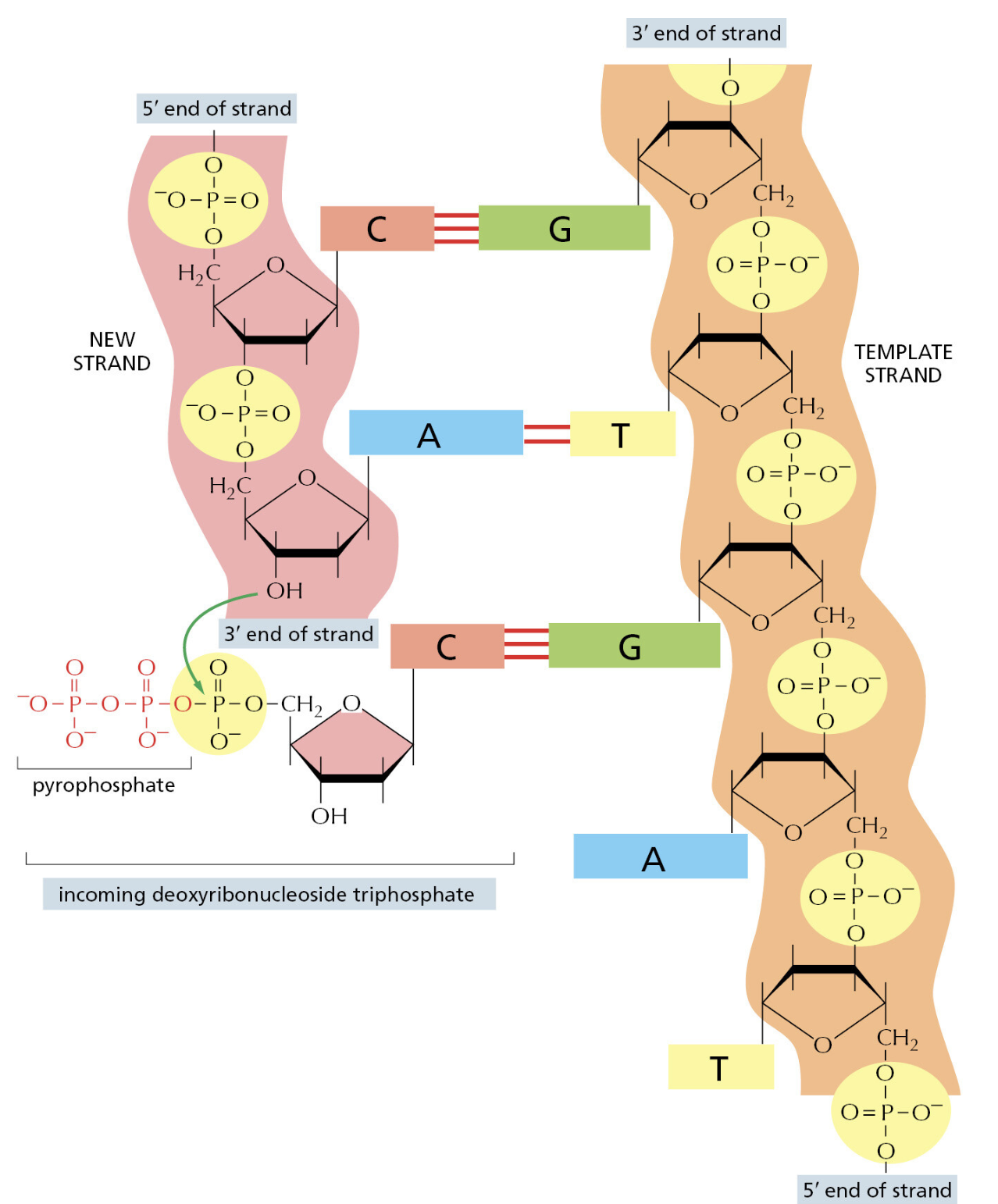
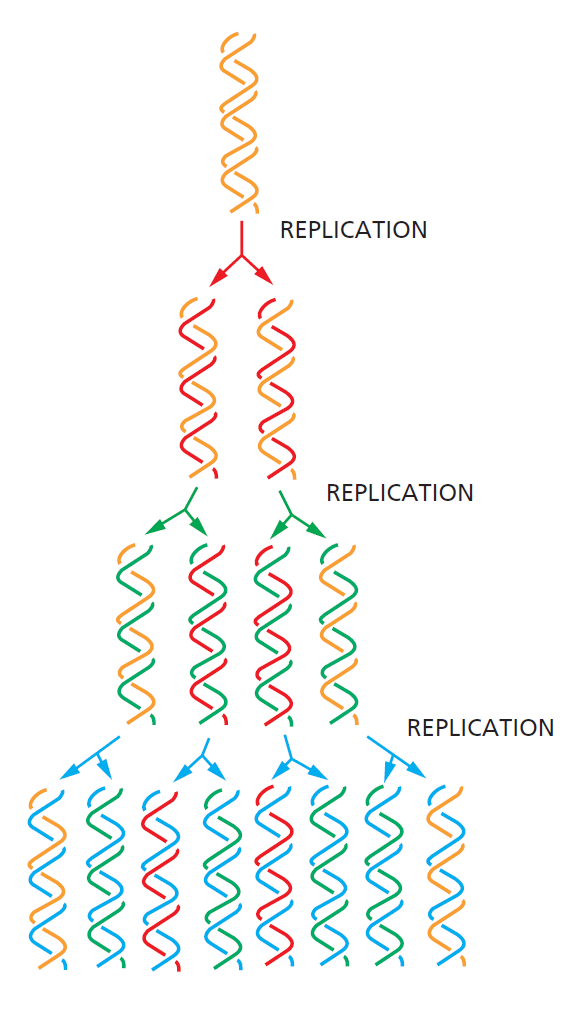
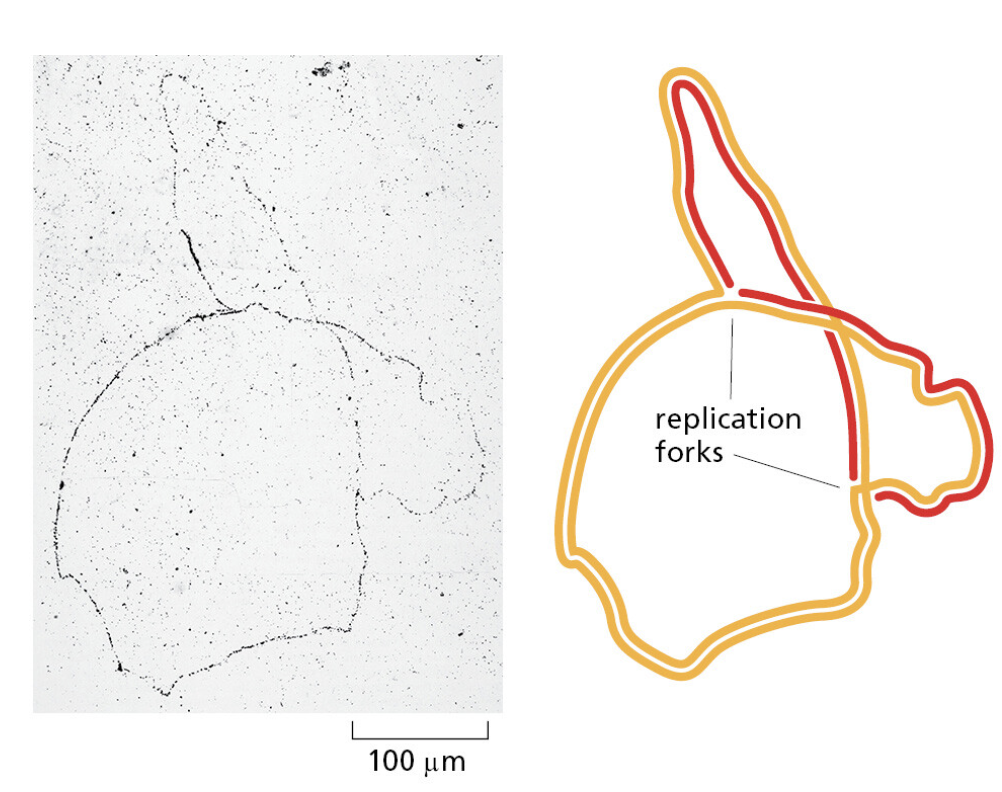
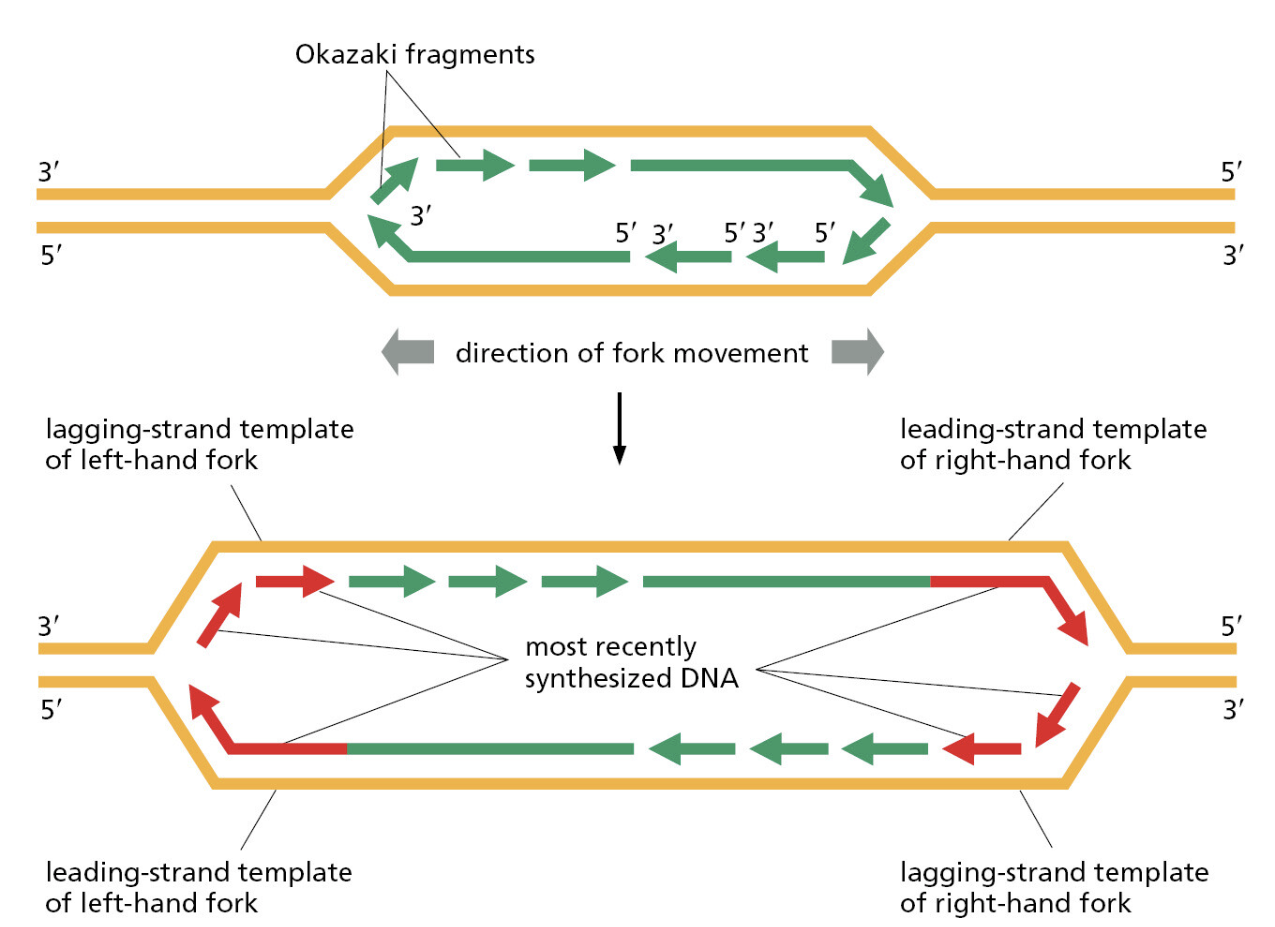


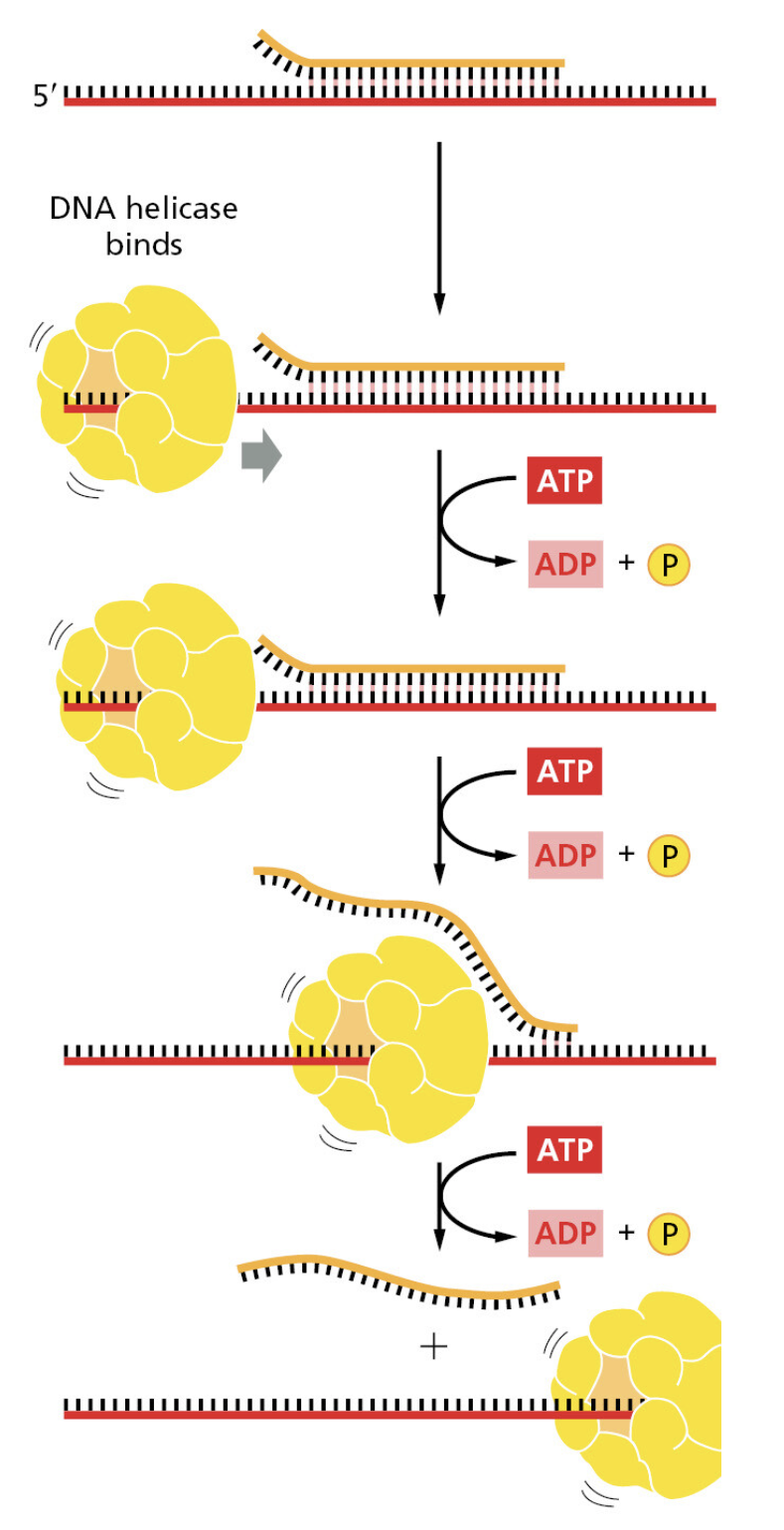
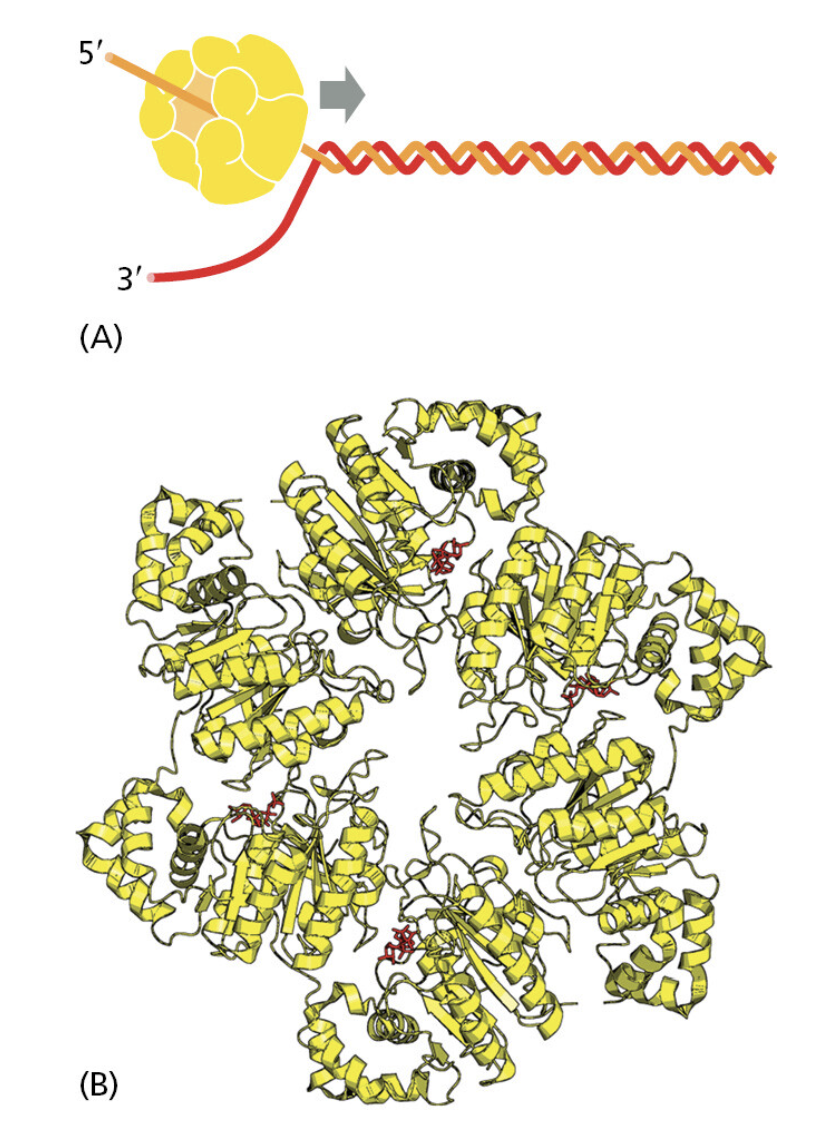
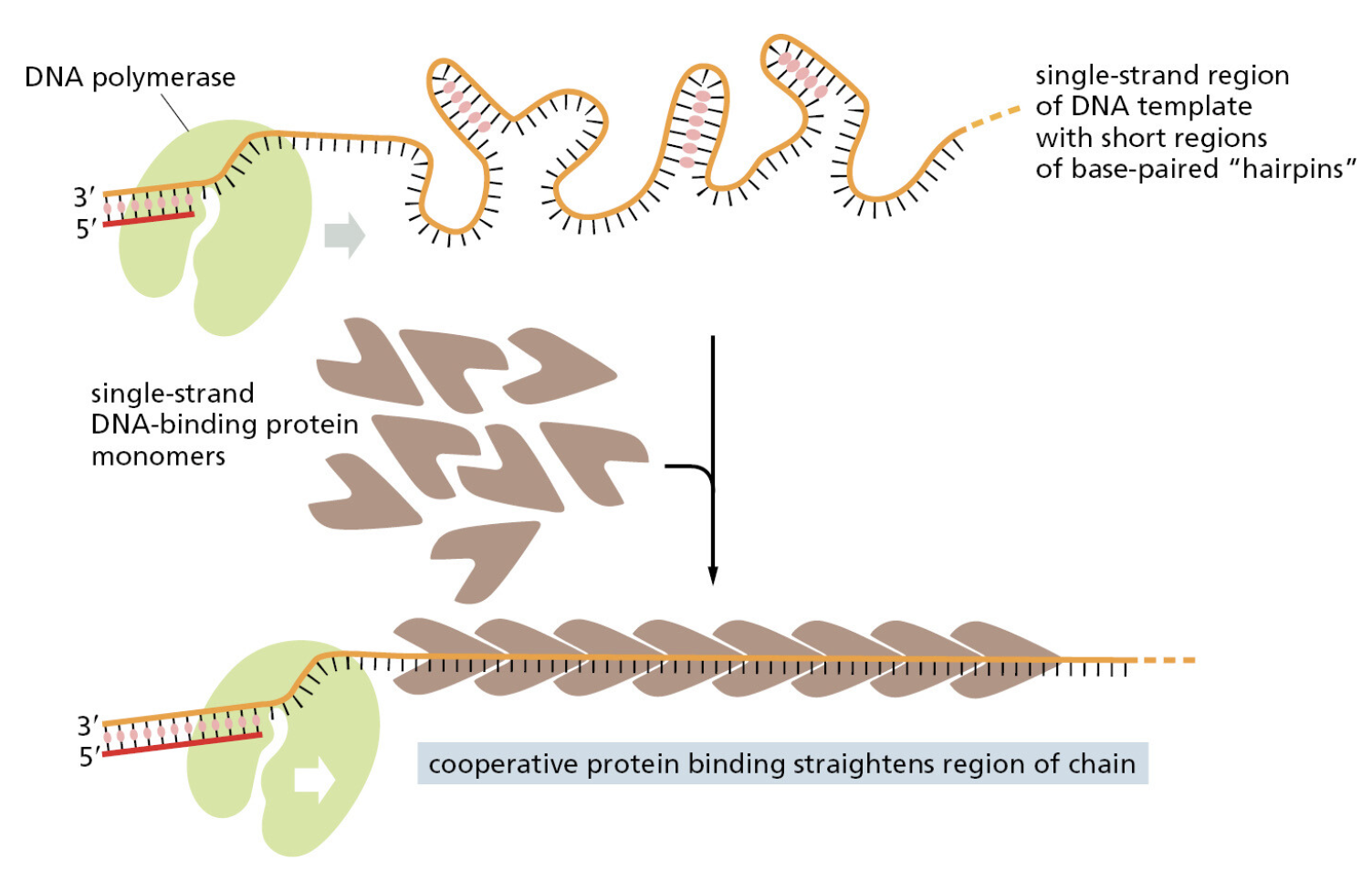
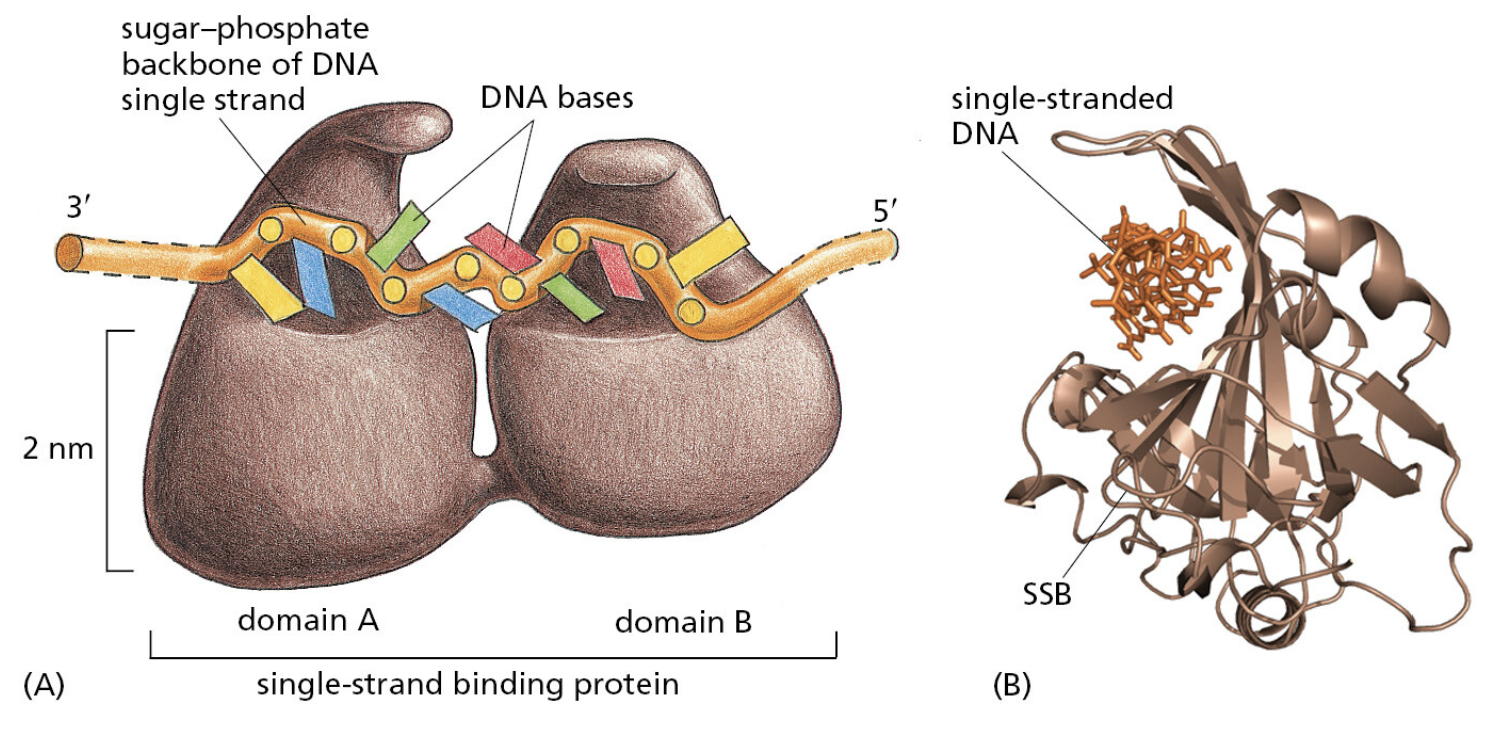
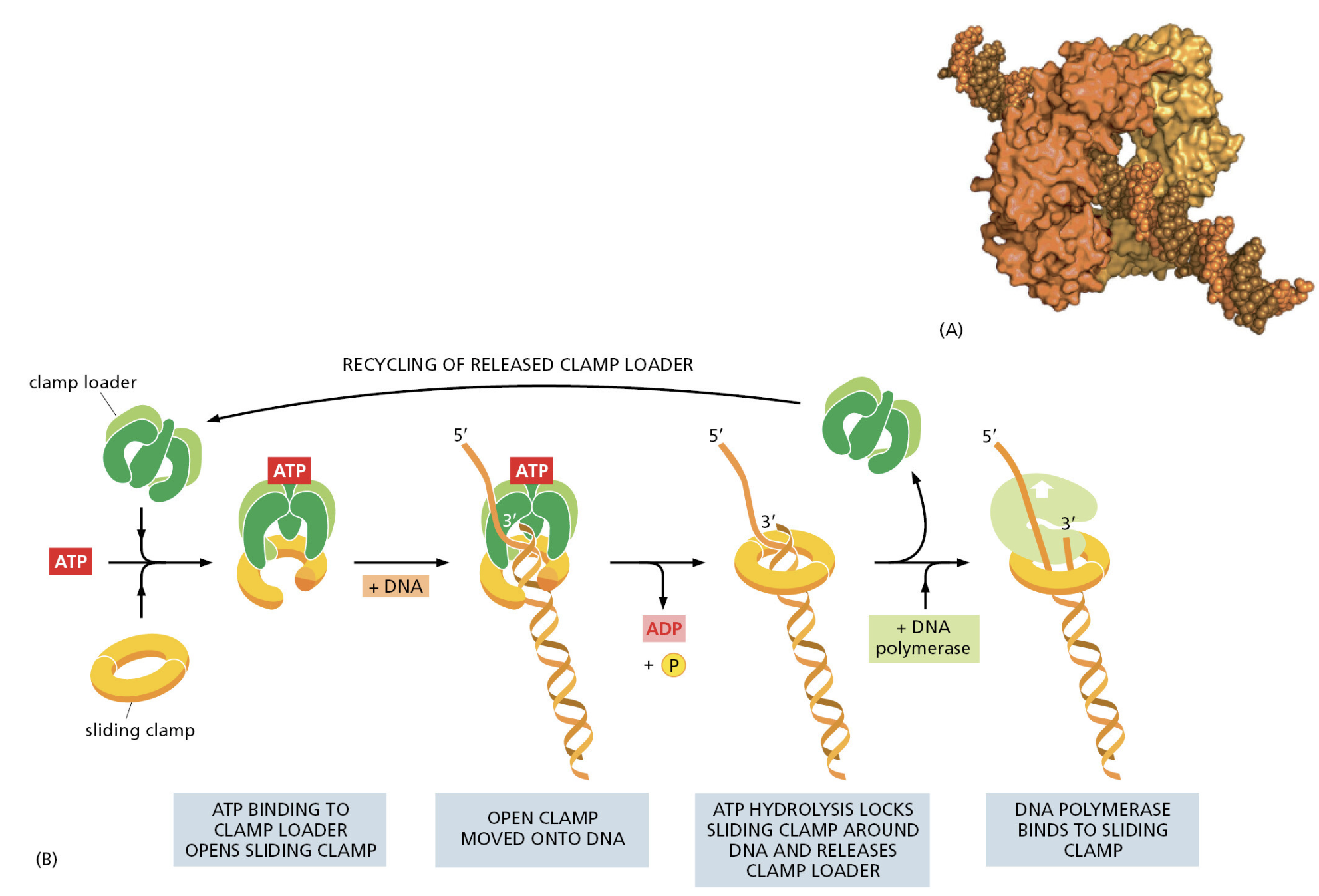
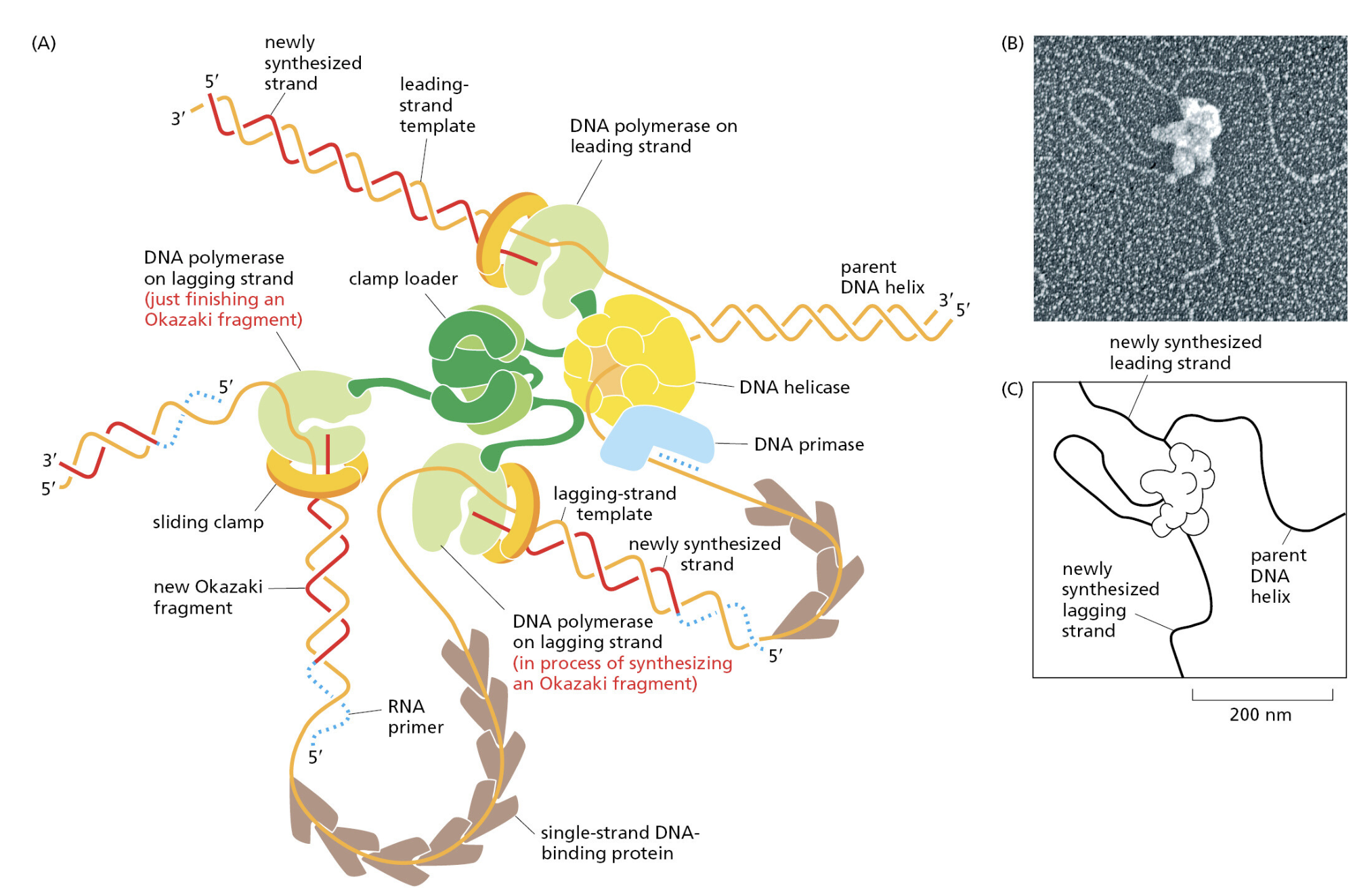

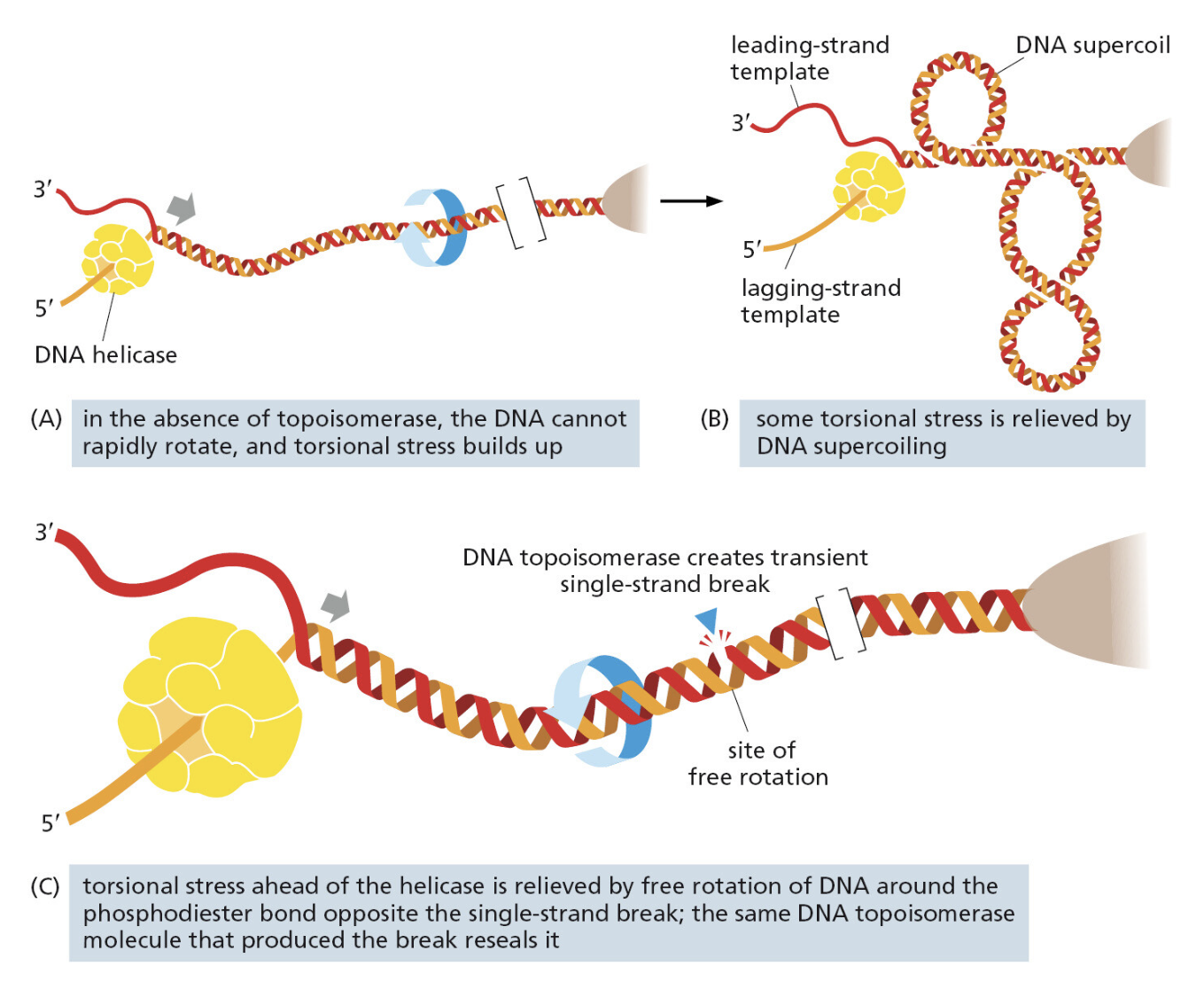
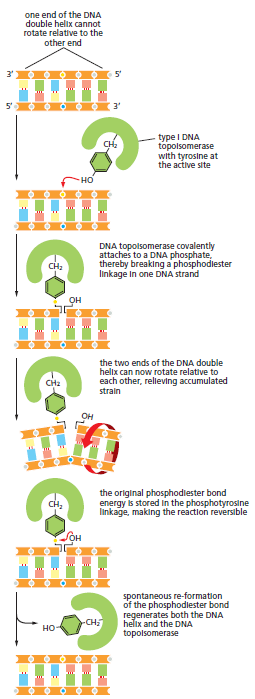
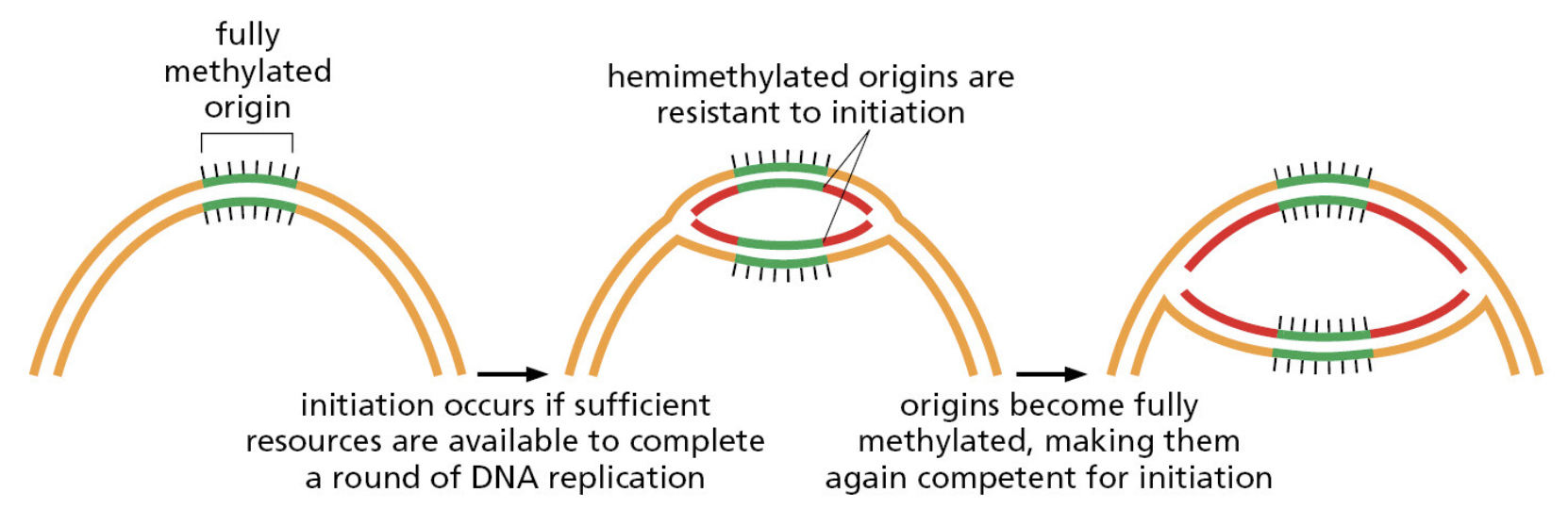
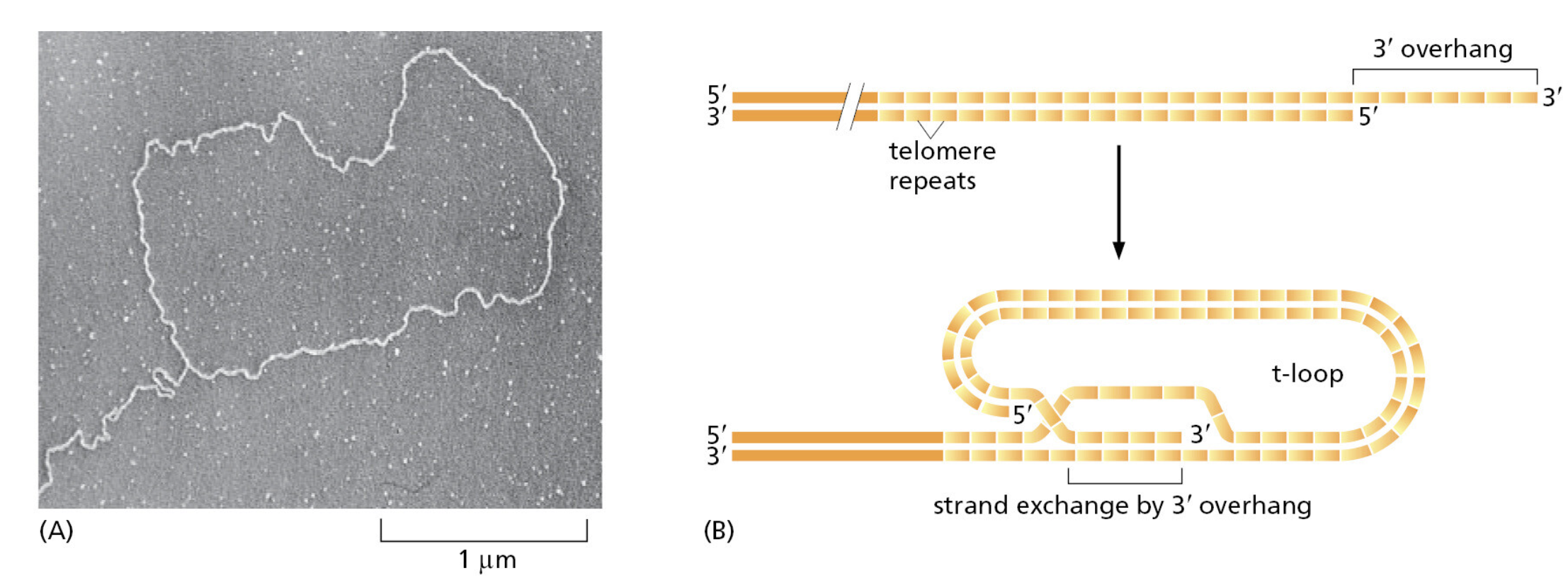
TTATCCACA)+ DUE(3个 13 bp 富 AT 重复)+ GATC 位点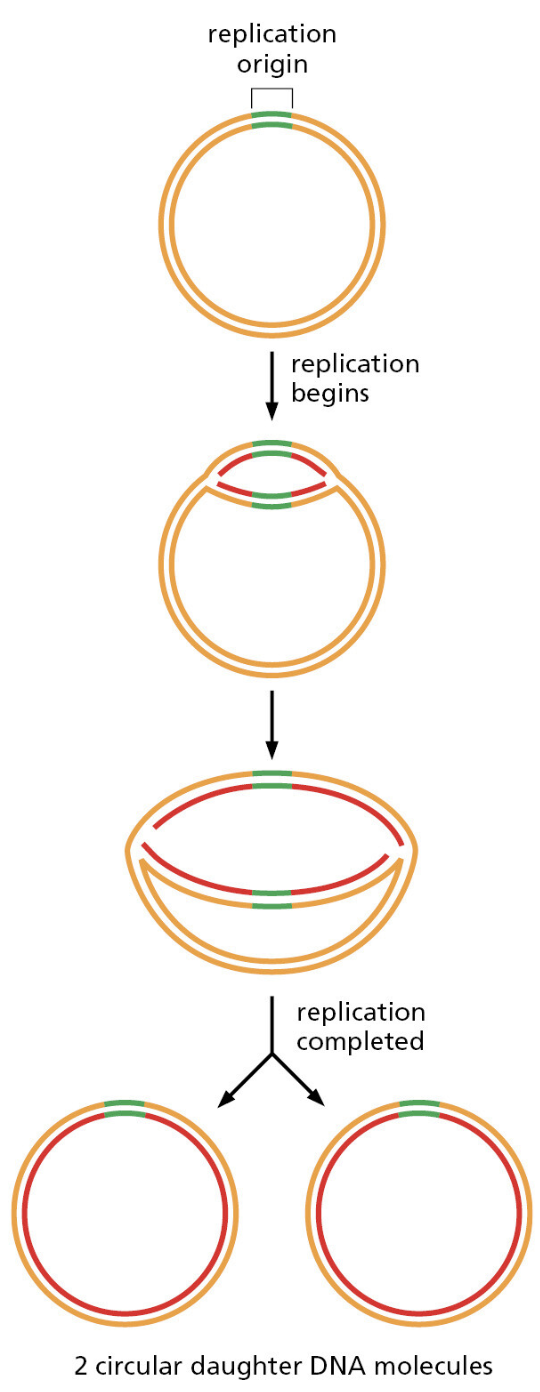

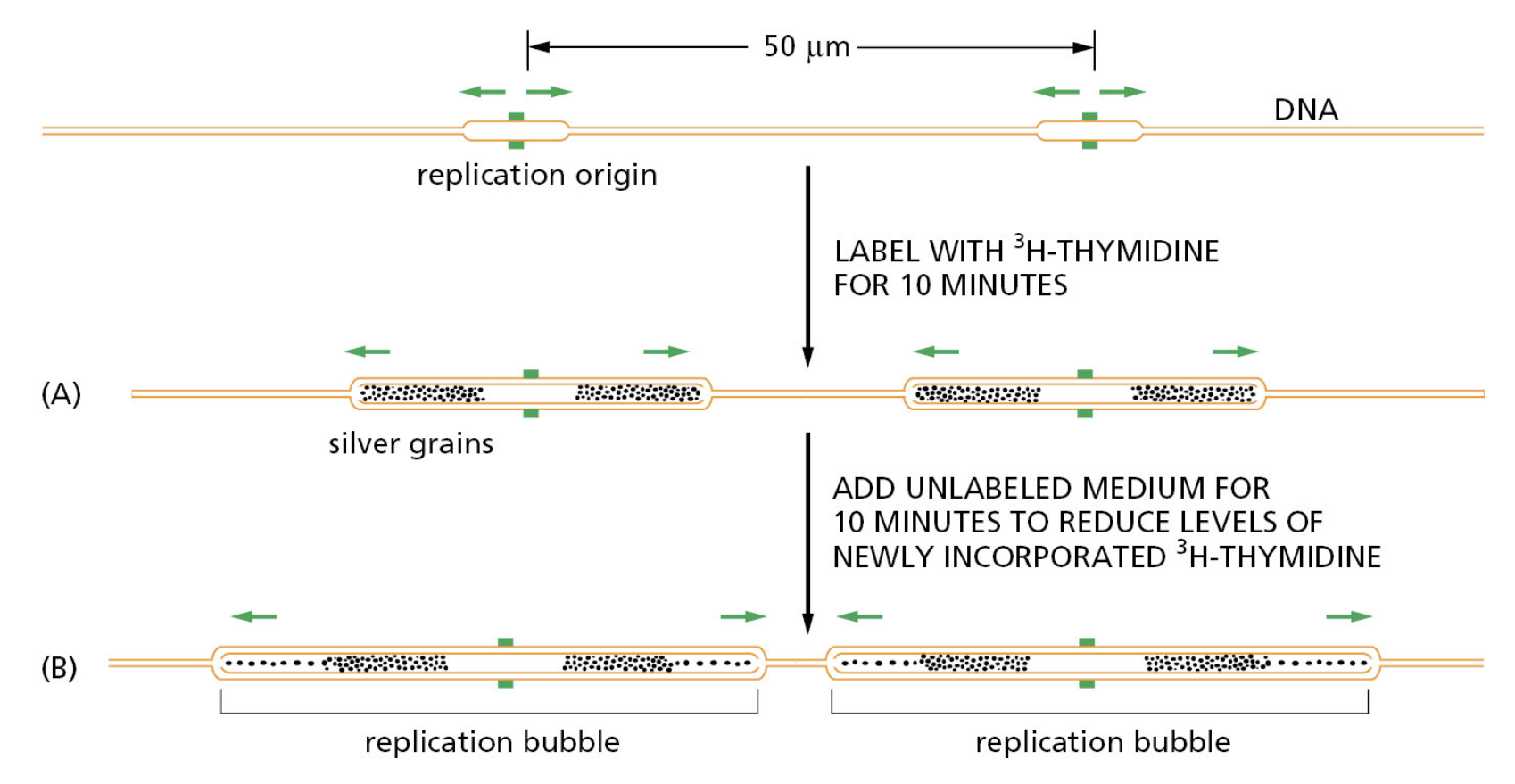
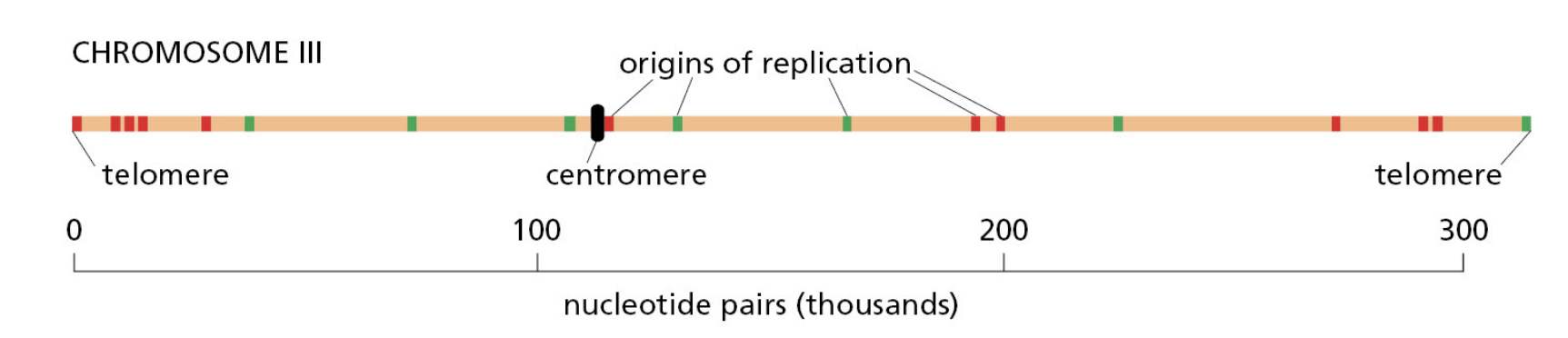
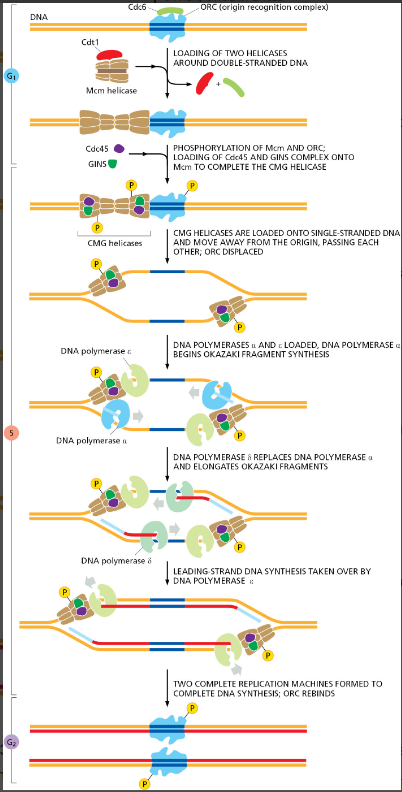

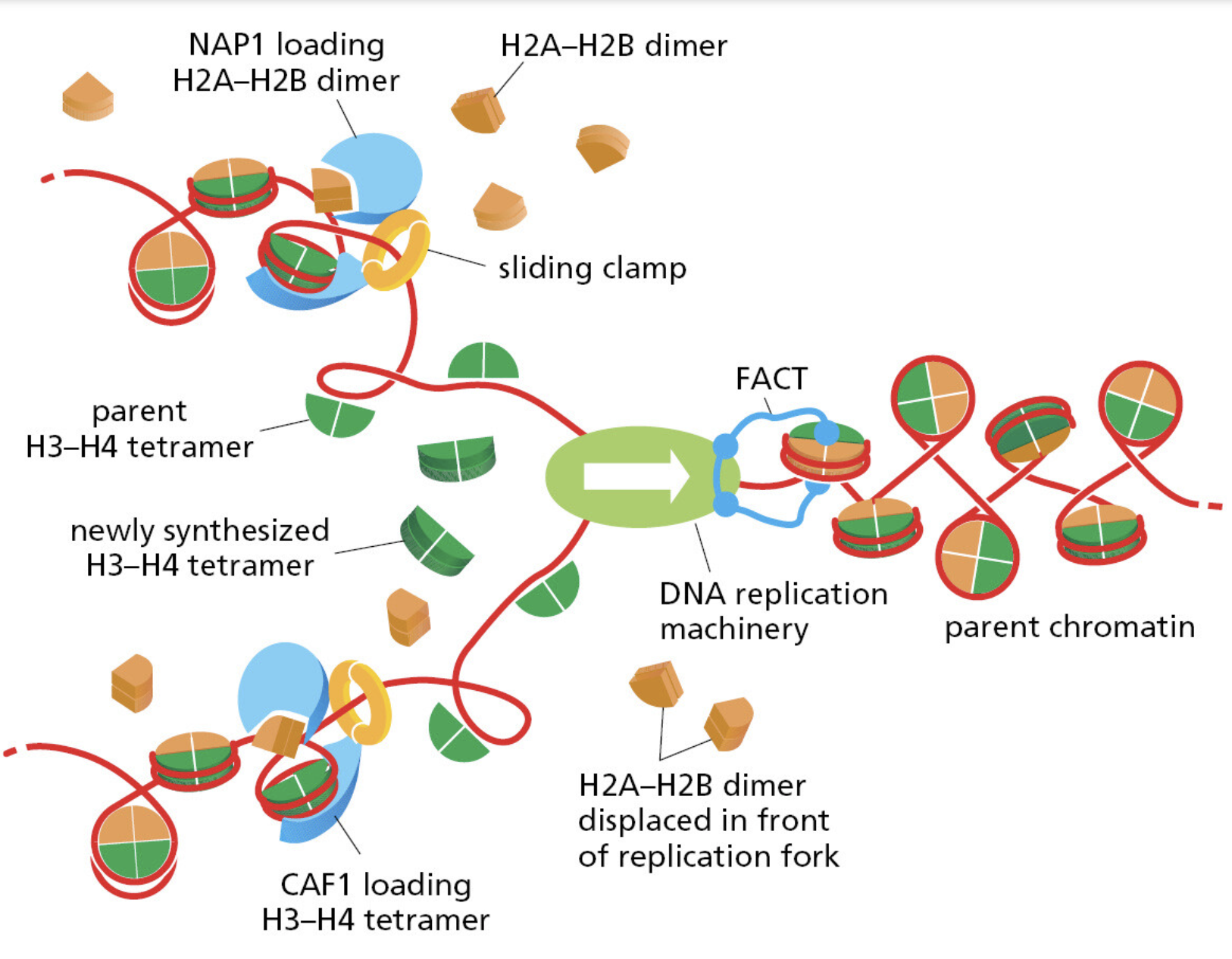
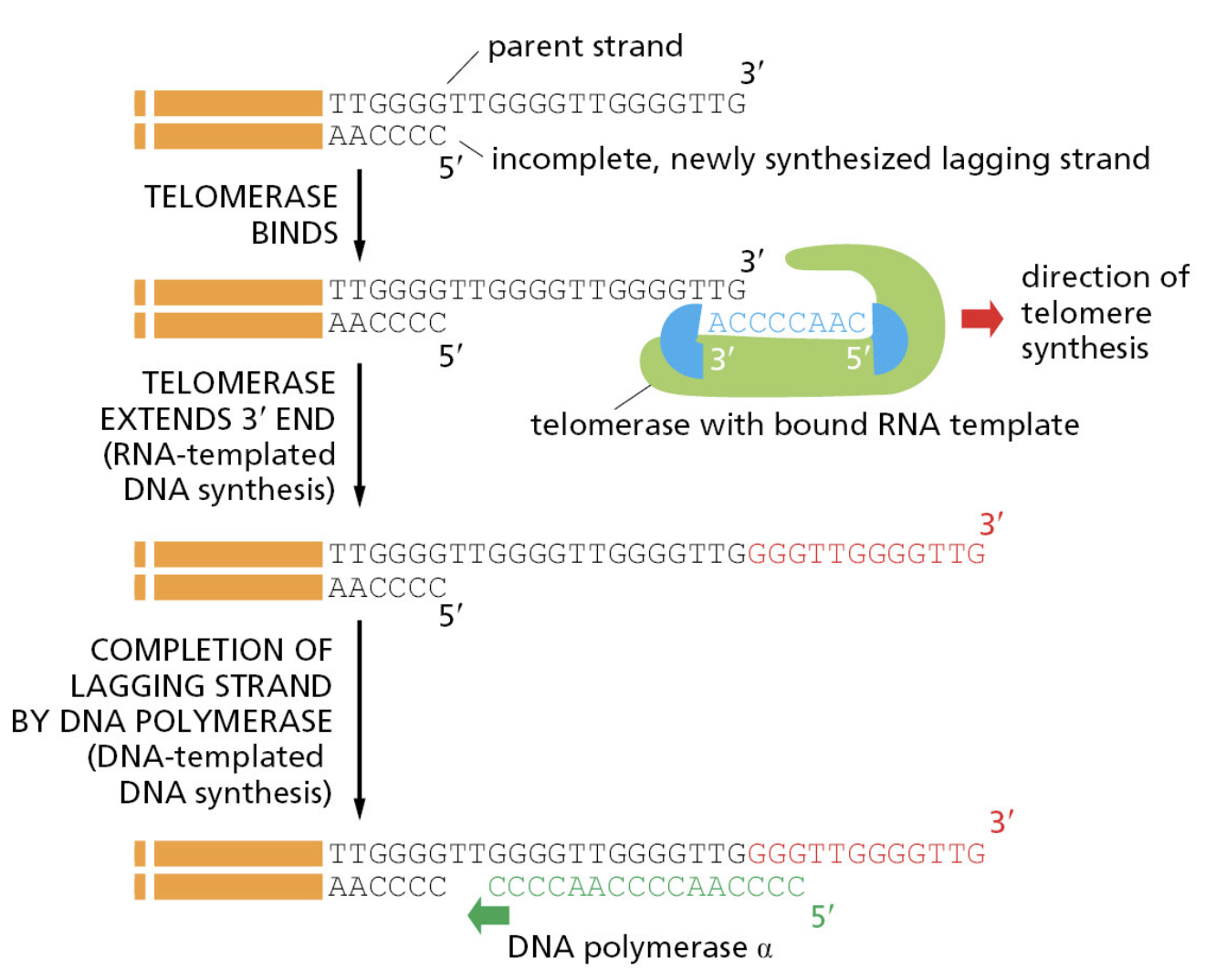
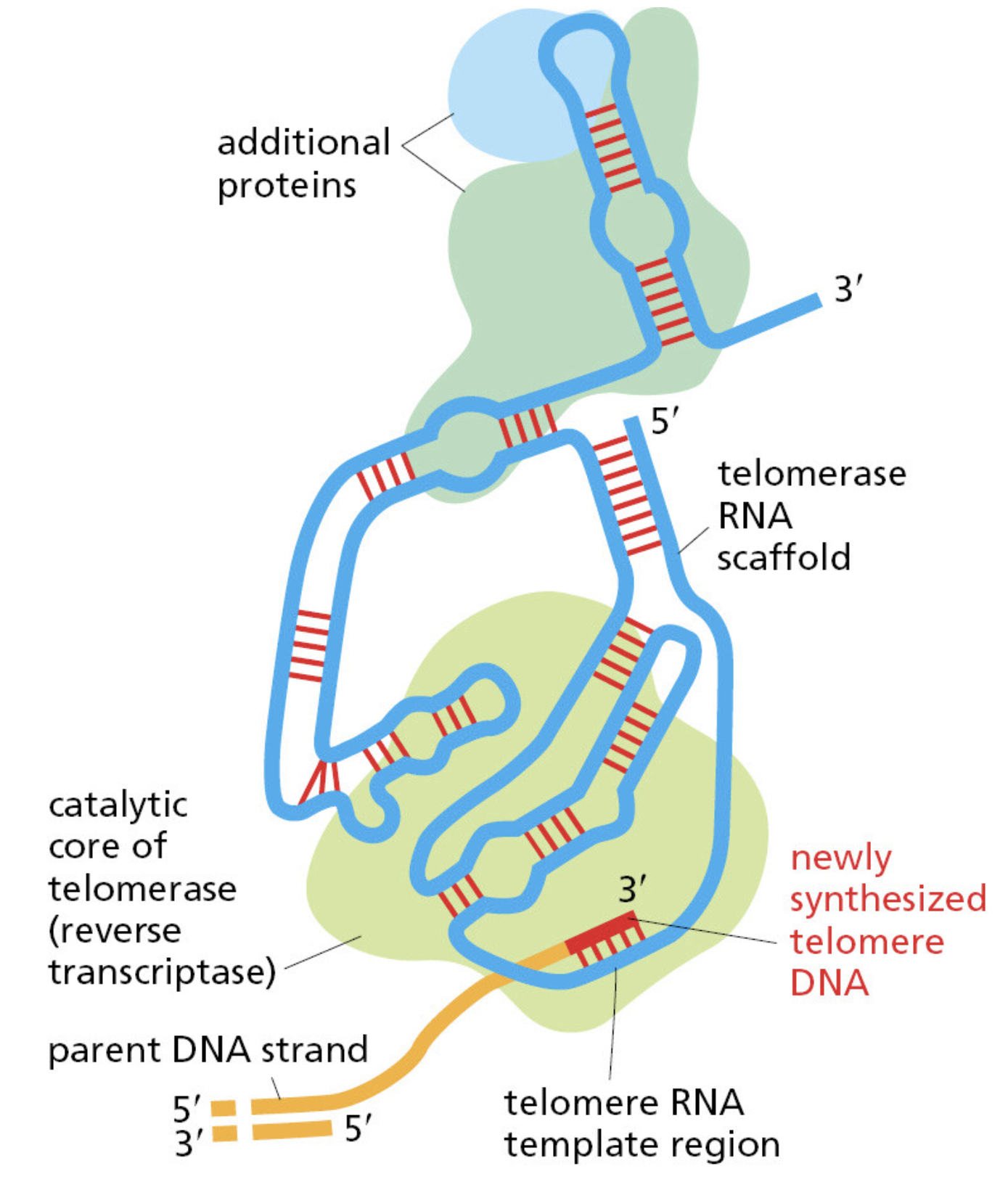
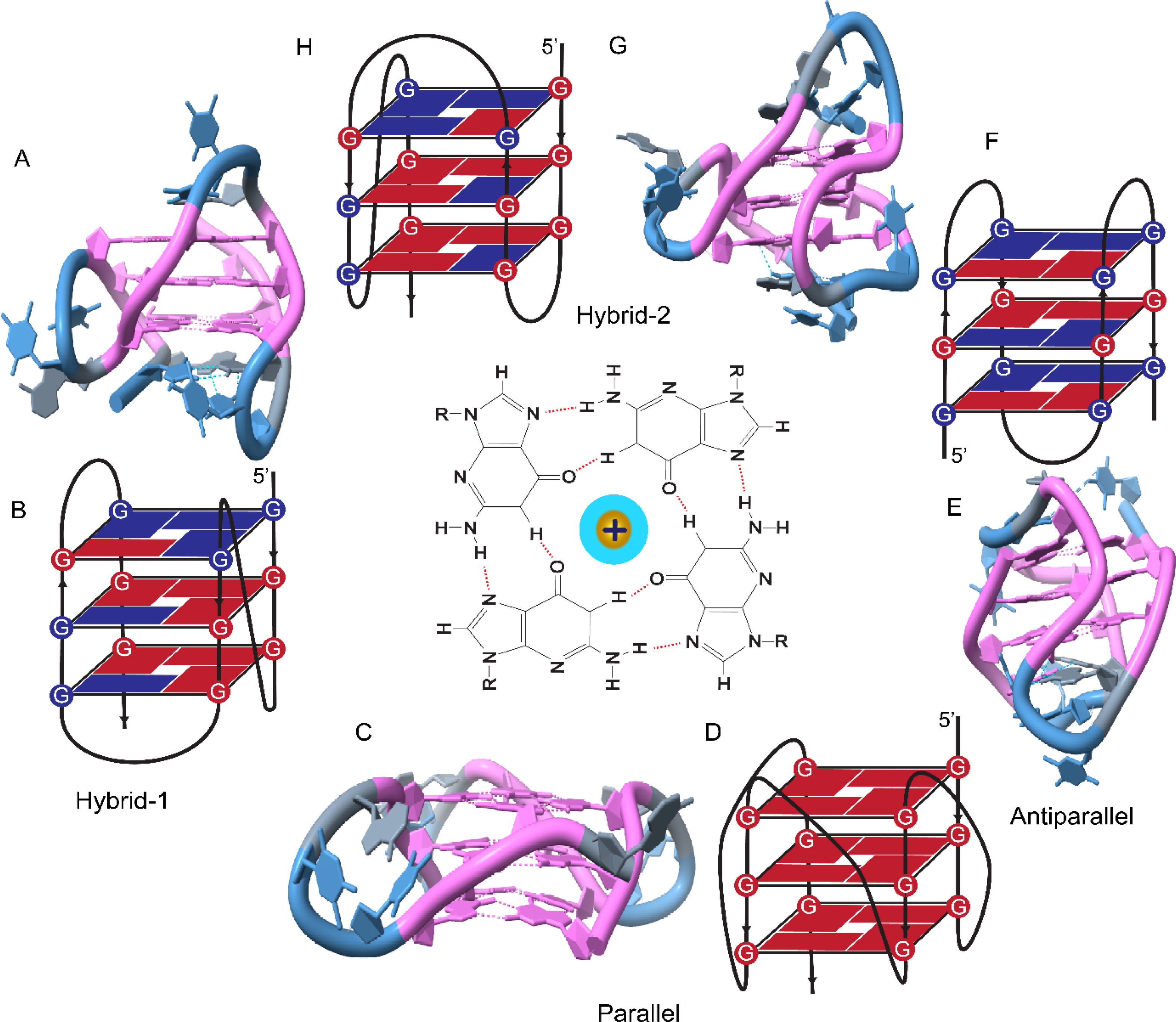
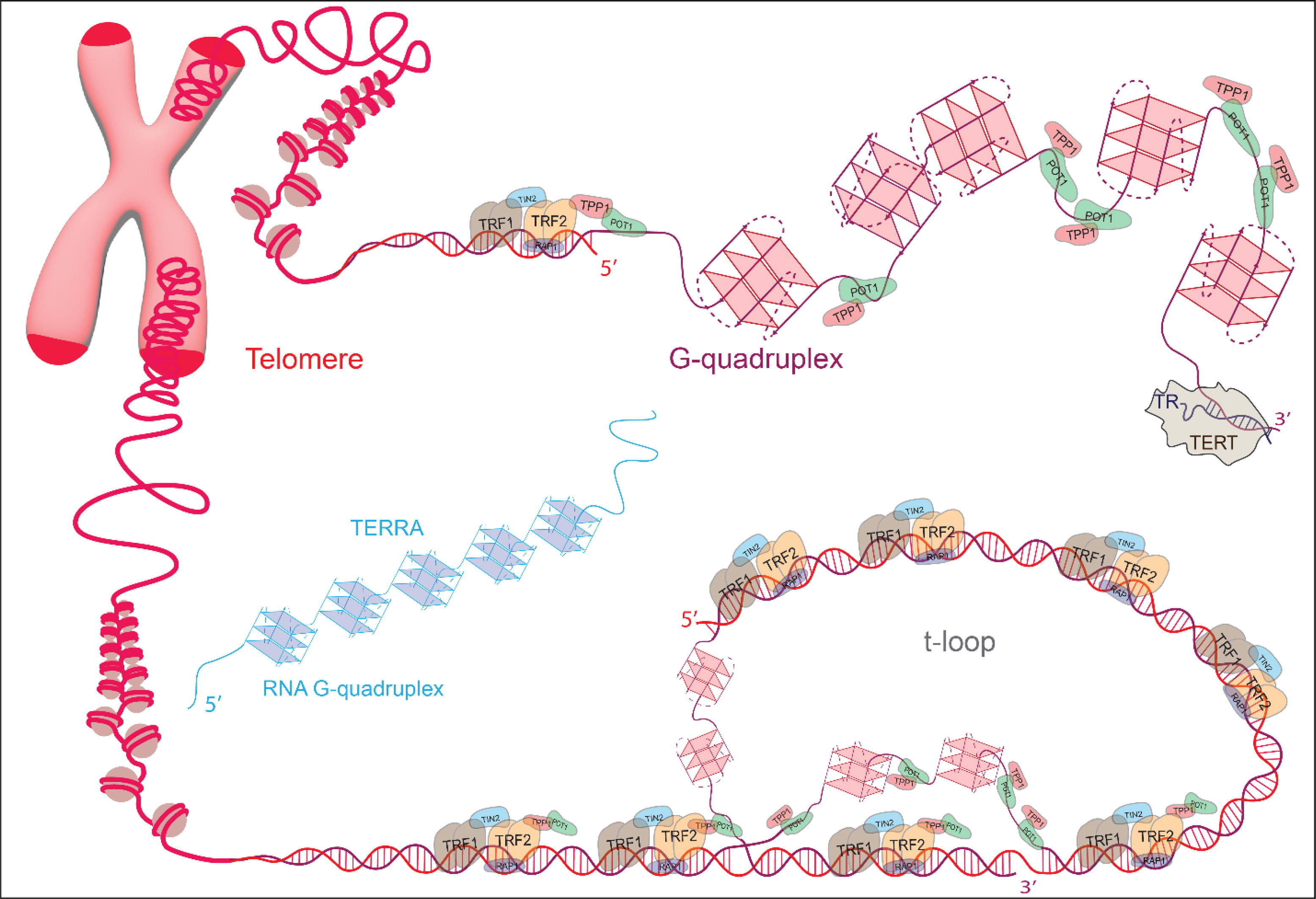

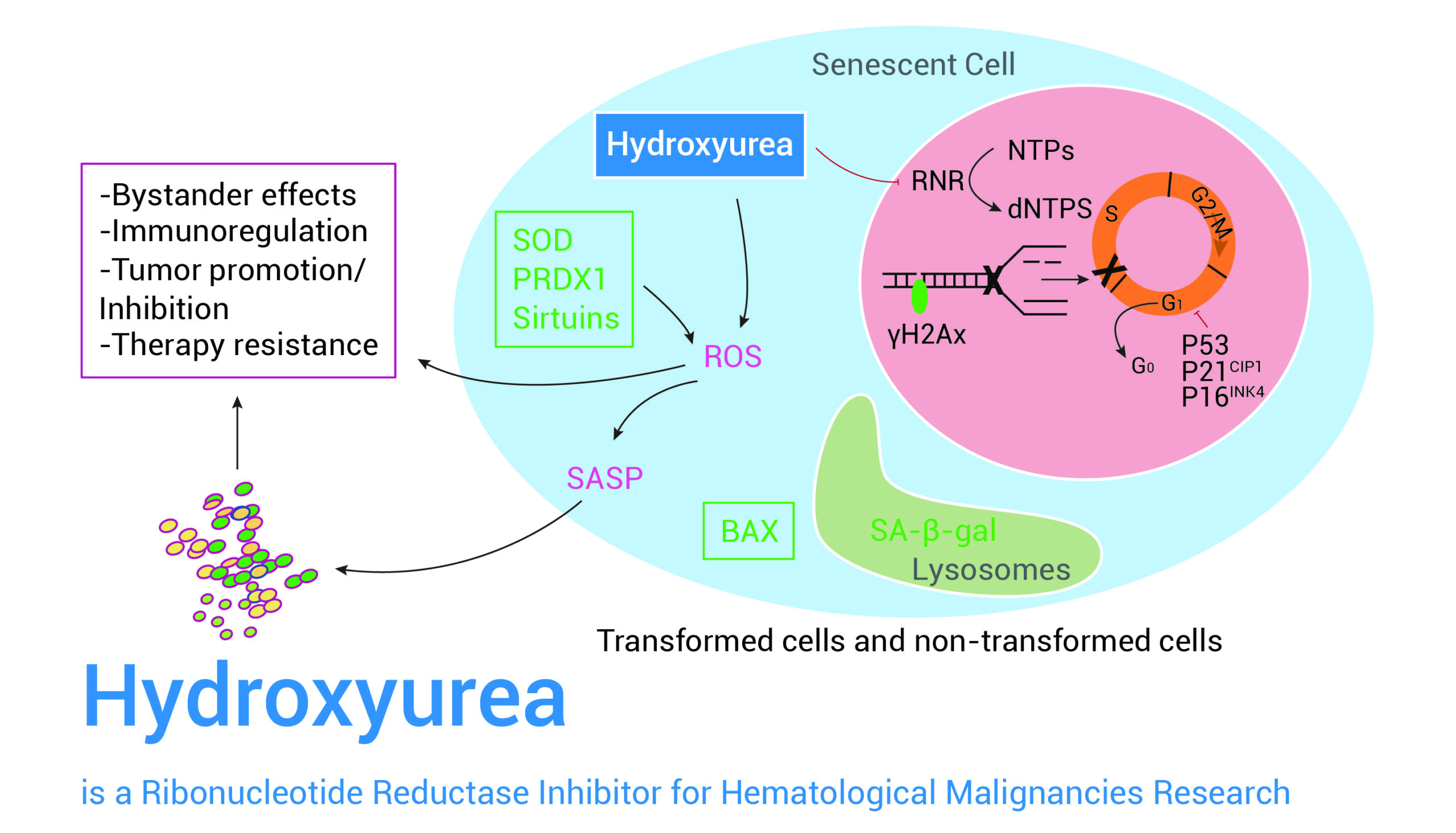
课堂问题:若单独抑制原料供给,哪些正常组织最容易出现副作用?
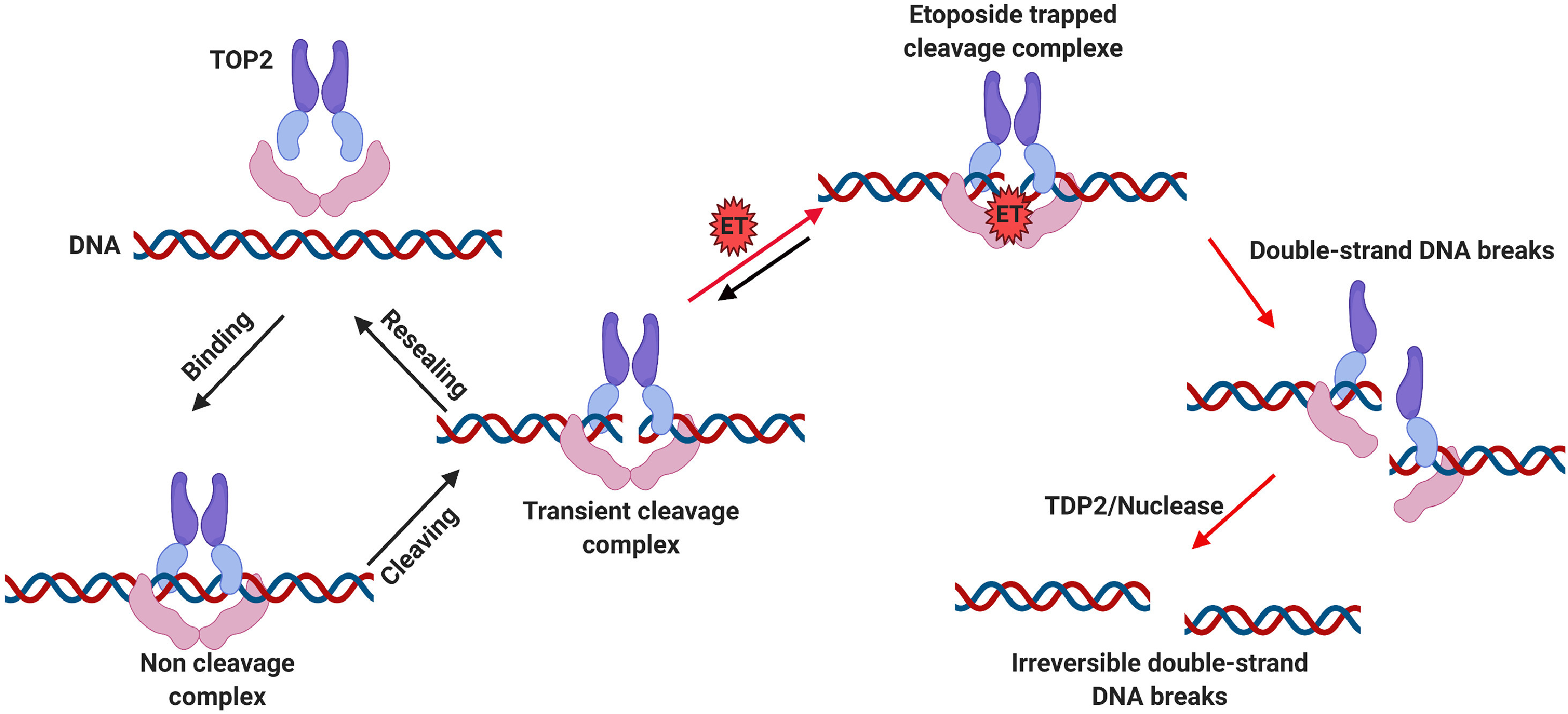
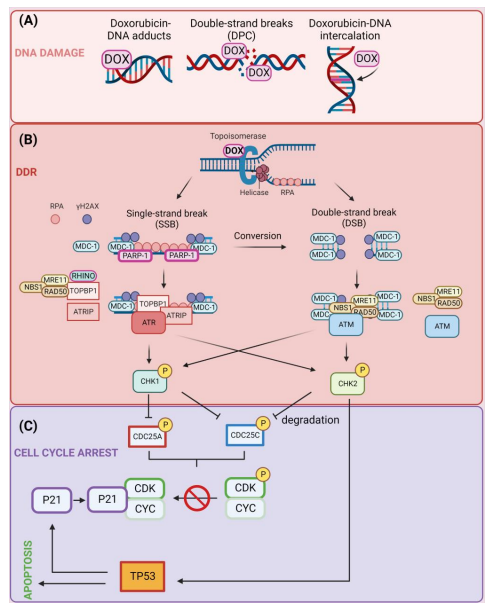
课堂问题:为什么高增殖肿瘤细胞更敏感,但骨髓和毛囊等正常组织也易受影响?
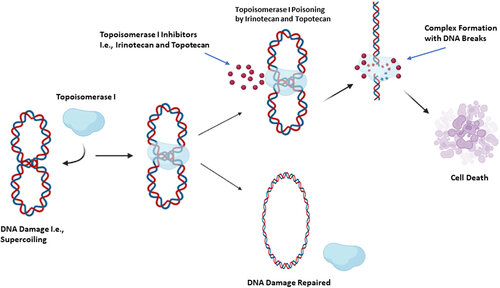
课堂问题:Topo I与Topo II抑制剂在机制和不良反应谱上会有哪些差别?
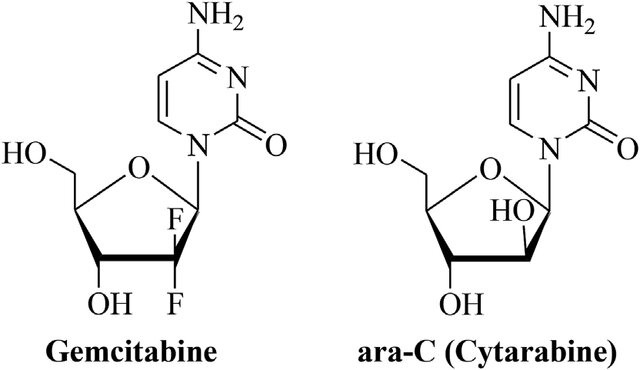
课堂问题:同为核苷类似物,为什么不同肿瘤对药物敏感性差异很大?
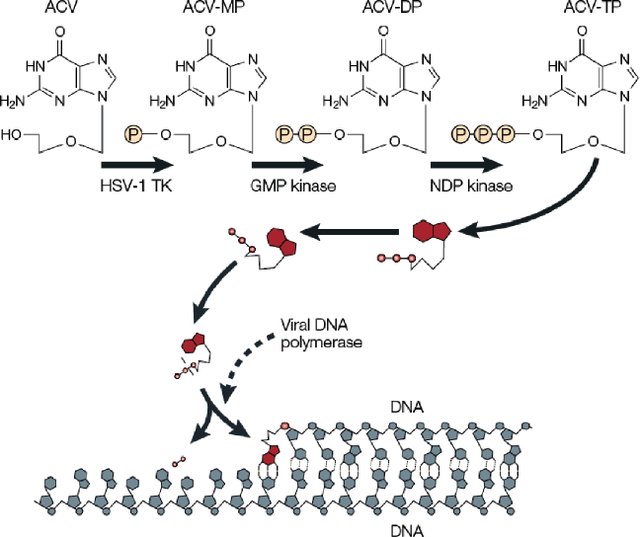
课堂问题:为何阿昔洛韦对不同病毒家族疗效差异明显?
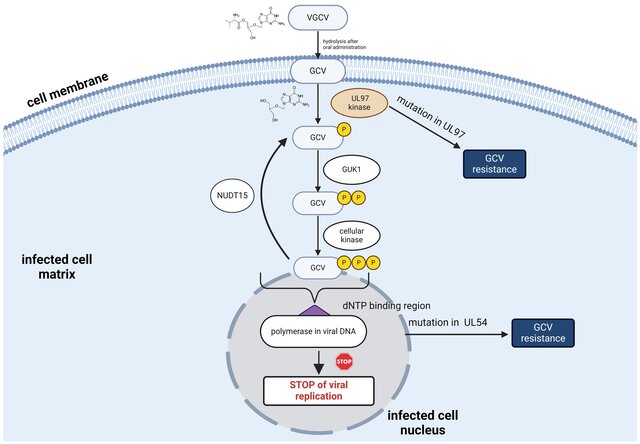
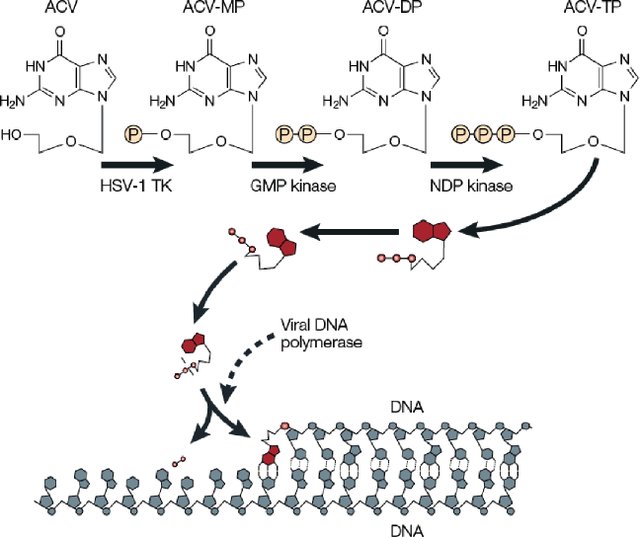
课堂问题:在重症或免疫抑制患者中,疗效与毒性如何做动态平衡?
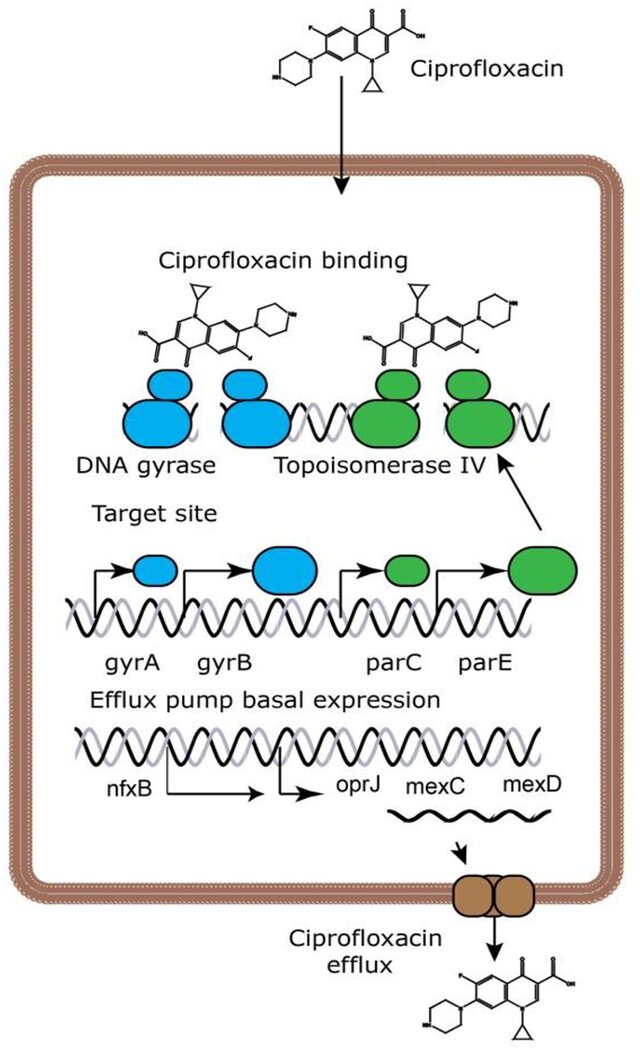
课堂问题:面对耐药上升,临床端和公共卫生端分别可以做什么?
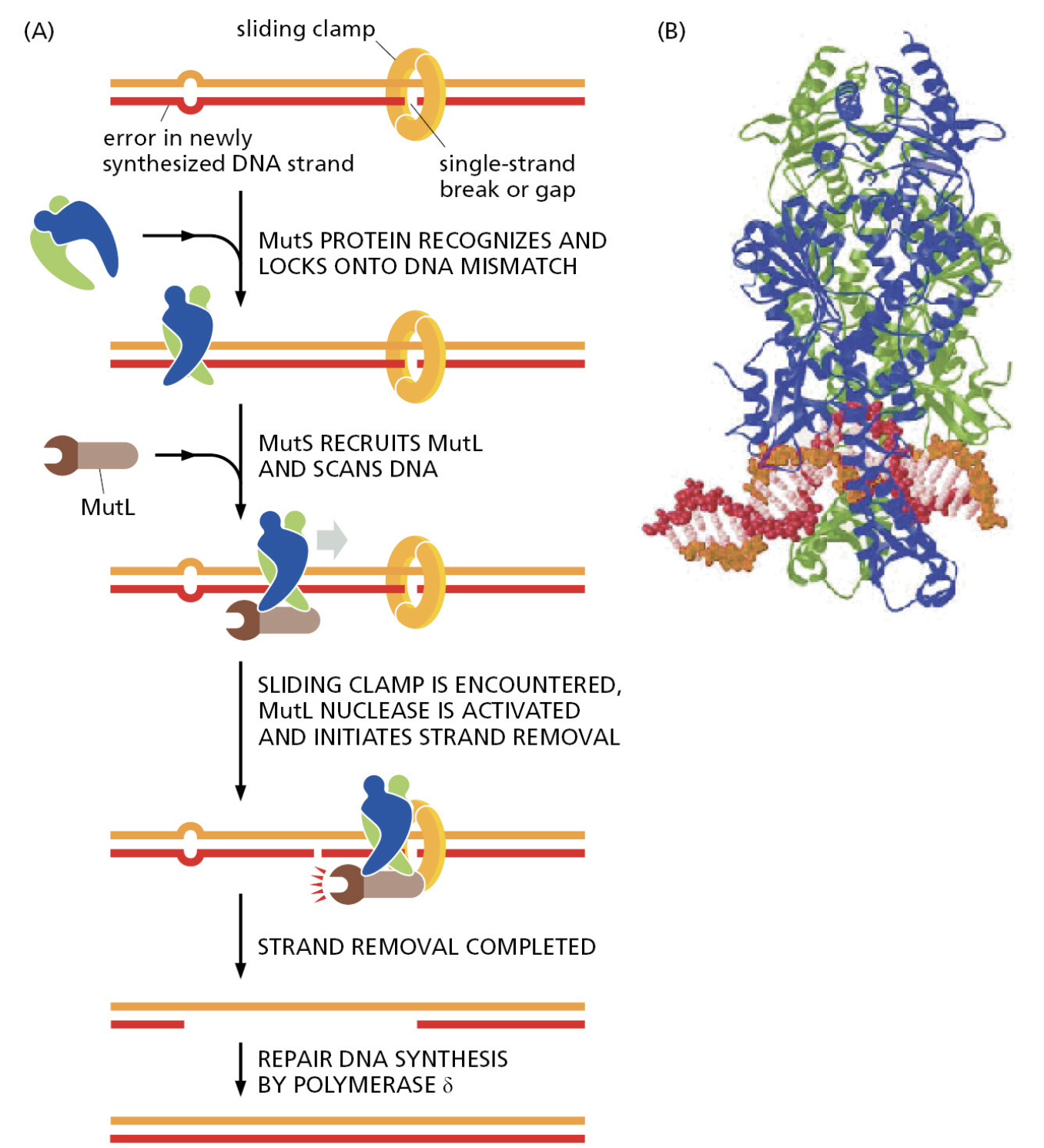
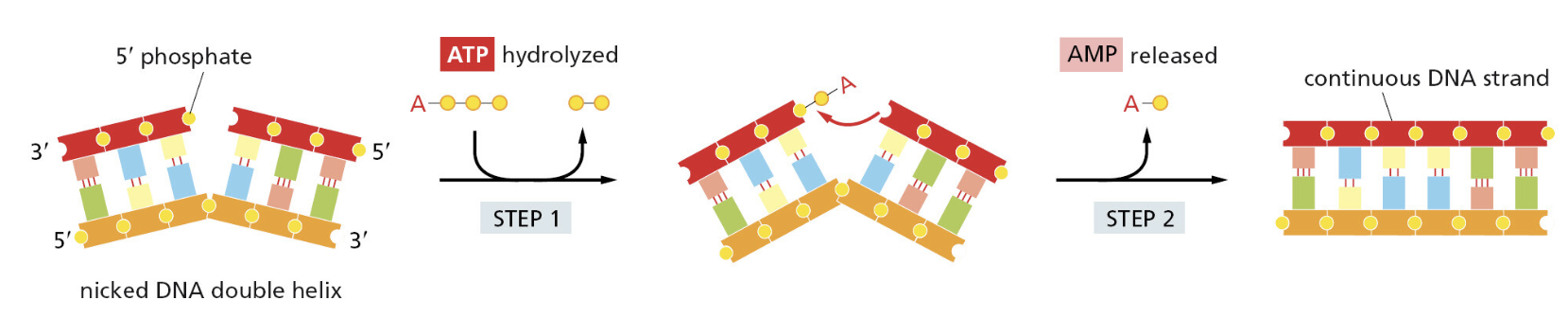
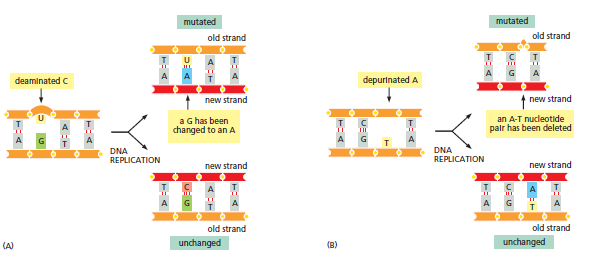
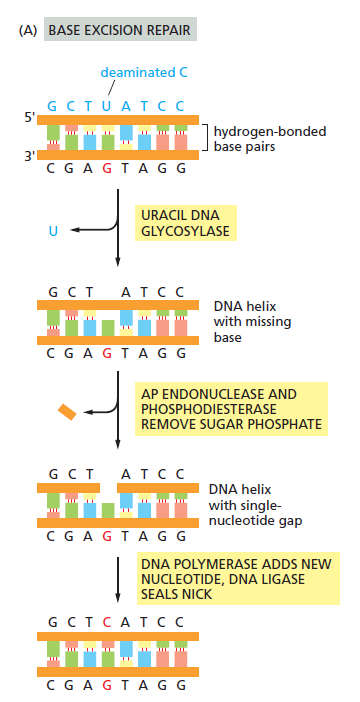
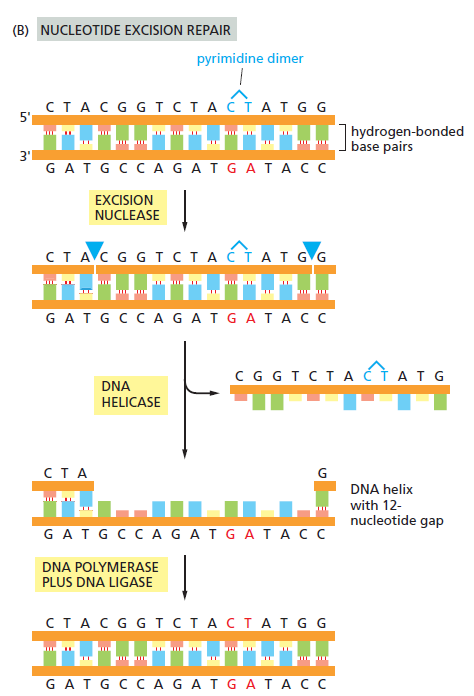
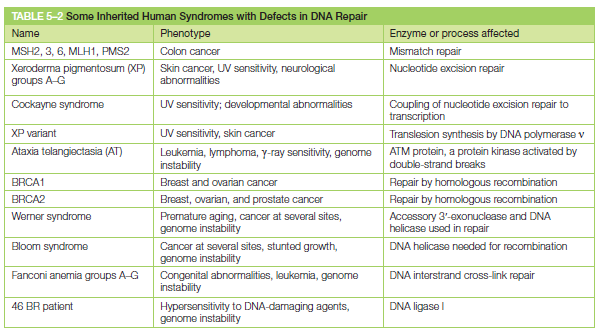
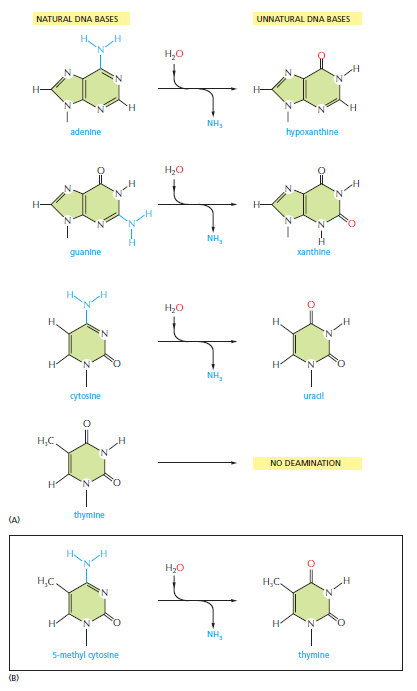
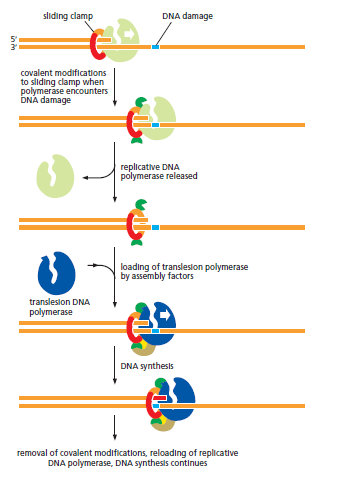
核心问题:细胞如何在如此高强度的化学攻击下仍精确维持基因组?答案就是本讲的主题——DNA 修复。
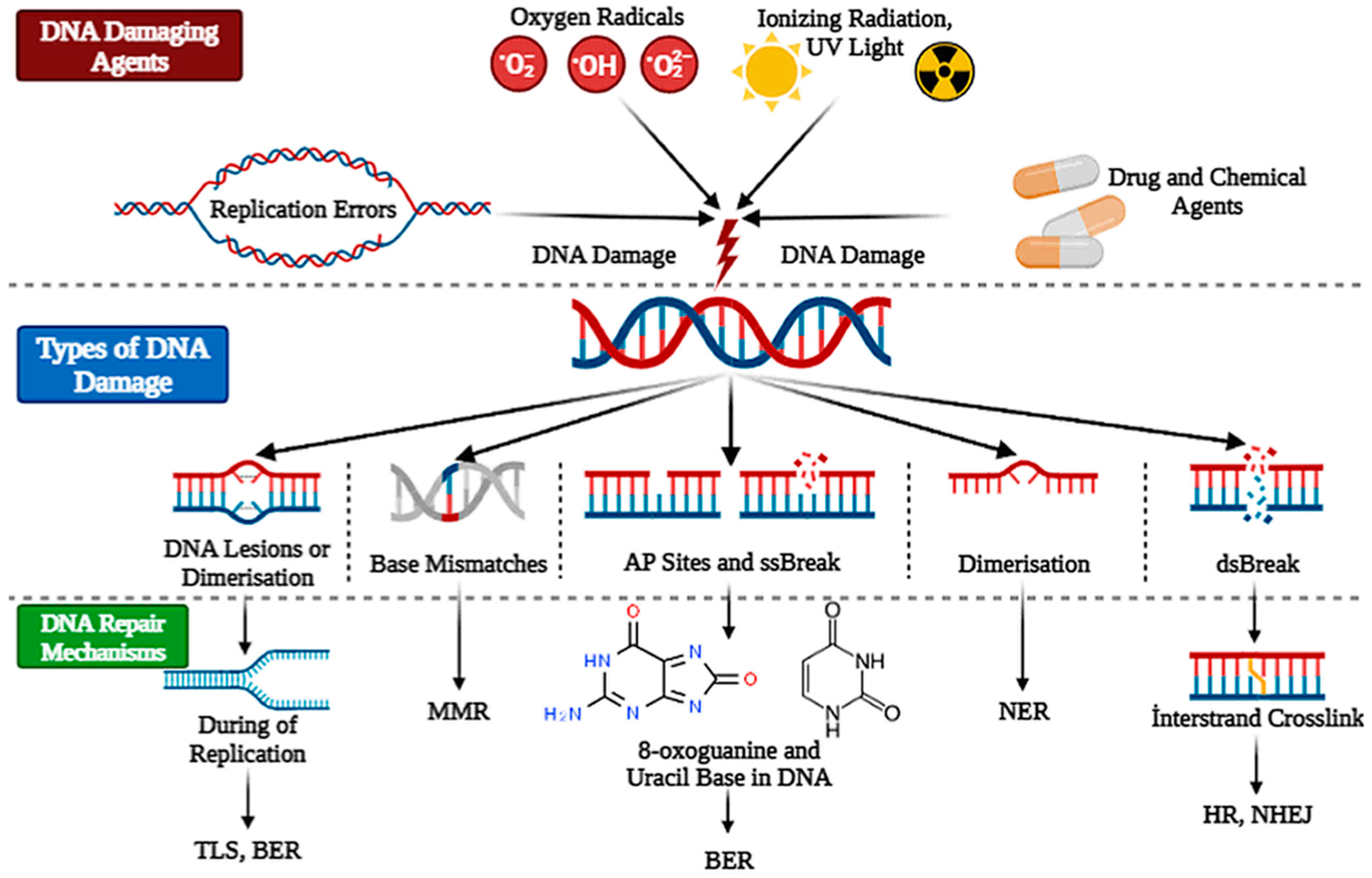
临床意义:理解修复通路直接推动了 PARP 抑制剂(合成致死策略)等靶向药物的开发——本讲后半段将详细介绍。
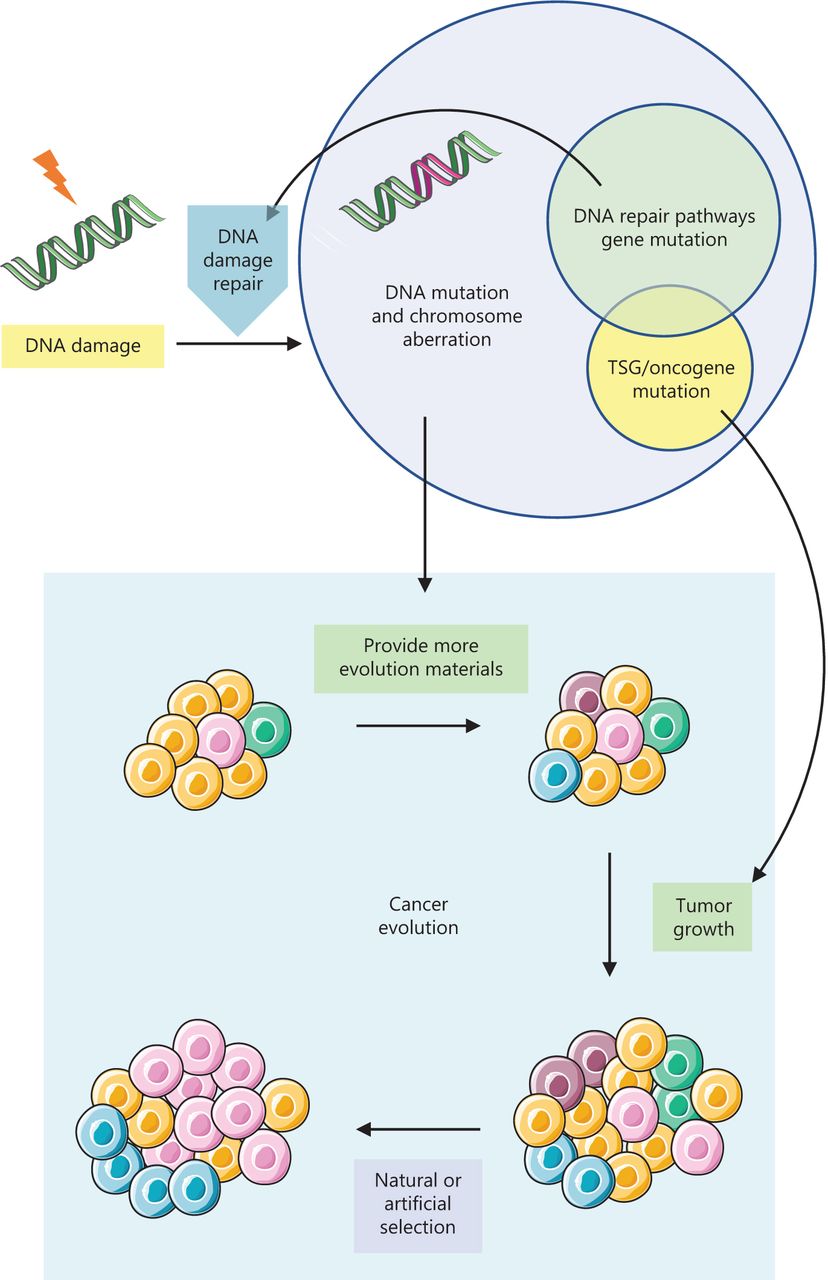
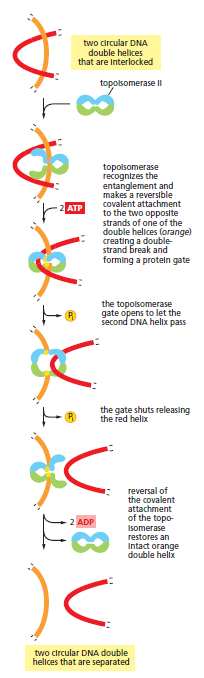
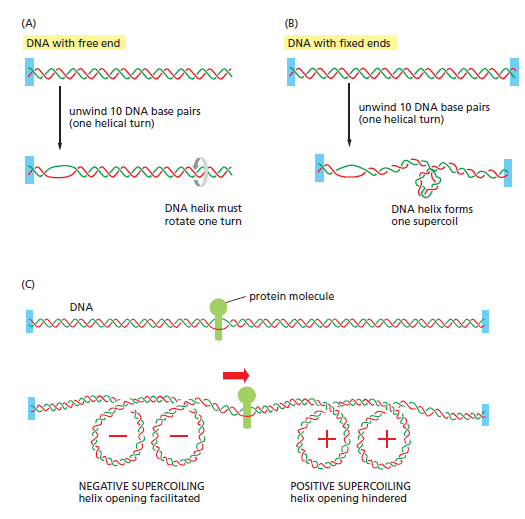
宏观视角:DNA 不是静态"蓝图",而是在修复、重组、转座的持续作用下不断演化的动态信息载体。本讲将从机制上揭示这三个过程。

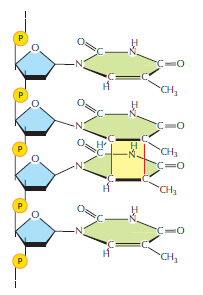

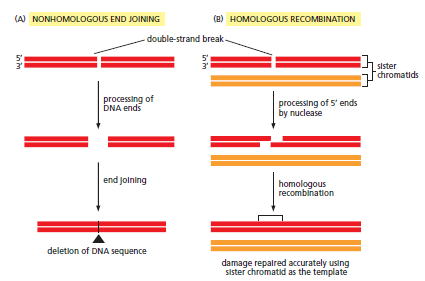
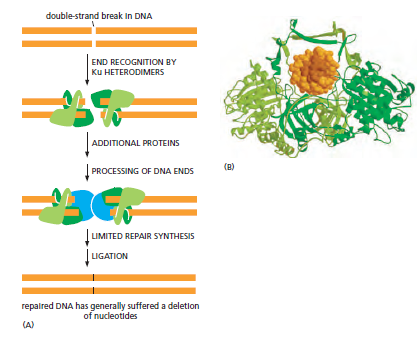


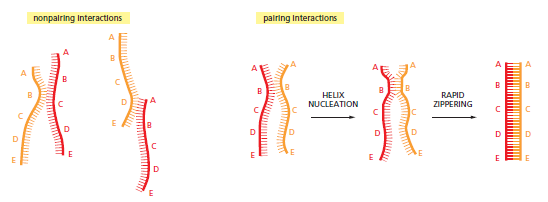
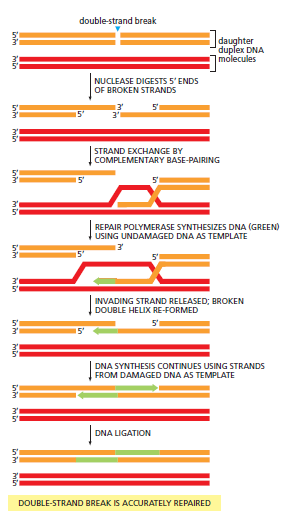
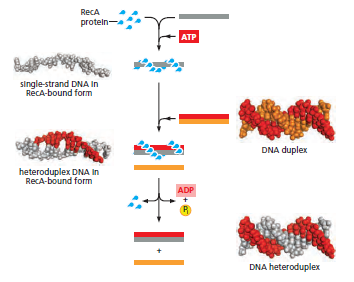
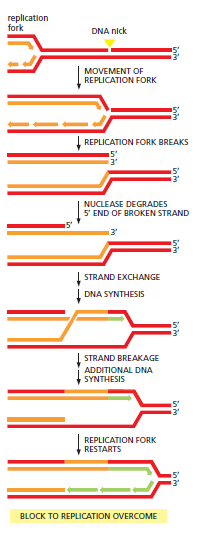
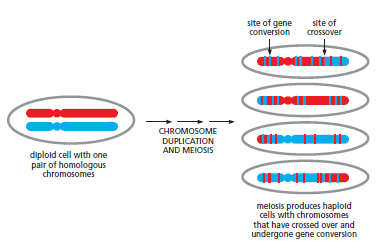
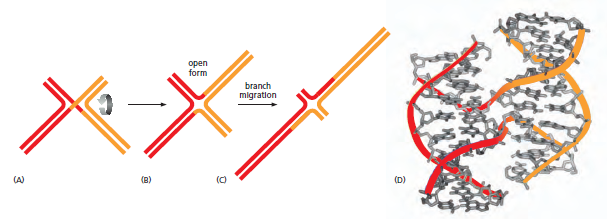
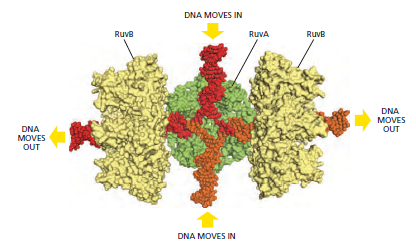
概念辨析:减数分裂 HR vs 体细胞 HR 修复(同一机制,两种用途)
| 维度 | 减数分裂中的同源重组 | 体细胞同源重组修复 |
|---|---|---|
| 触发 | Spo11 主动制造 DSB(程序性) | 偶发 DNA 损伤(被动响应) |
| 配对模板 | 同源染色体(父-母两条) | 姐妹染色单体(自我克隆) |
| 时机 | 减数分裂前期 I(pachytene) | S / G2 期(必须有姐妹染色单体) |
| 频率 | 每条染色体强制 ≥1 次(保证正确分离) | 损伤事件触发,按需进行 |
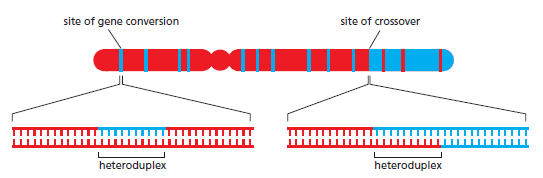
| 主要结果 | 交叉互换 + 基因转换 → 遗传多样性 | 精确复原原始序列 → 无 LOH |
| 关键蛋白 | Spo11、Dmc1(减数分裂特异)+ Rad51 | Rad51、BRCA1 / BRCA2、RPA、MRN |
| 失败后果 | 染色体不分离 → 三体 / 不育 | DSB 累积 → 凋亡 / 染色体不稳定 / 癌症 |
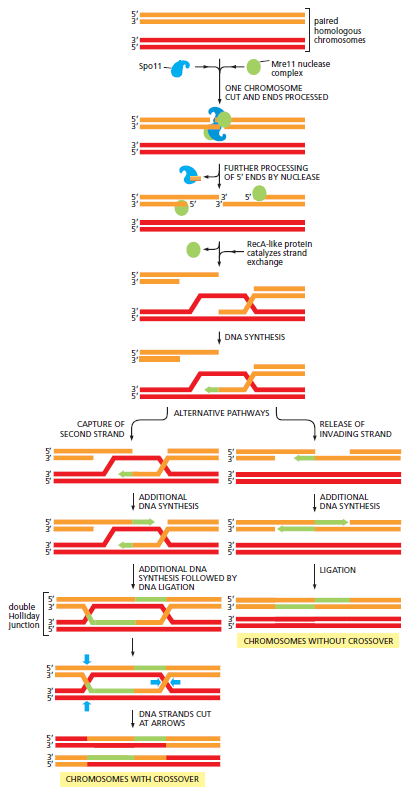
概念辨析:同源染色体 vs 姐妹染色单体
| 维度 | 同源染色体(homologs) | 姐妹染色单体(sister chromatids) |
|---|---|---|
| 来源 | 一条来自母方、一条来自父方 | S 期复制同一条染色体产生的两份拷贝 |
| 等位基因 | 基因座相同,等位基因可能不同(如 A vs a) | 完全相同(仅复制错误造成极个别差异) |
| 物理结构 | 独立两条染色体,平时不相连 | 在着丝粒处相连,呈 X 形 |
| HR 模板用途 | 减数分裂中故意使用 → 交叉互换 → 遗传多样性 | 体细胞 HR 修复的标准模板 → 精确复原,避免 LOH |
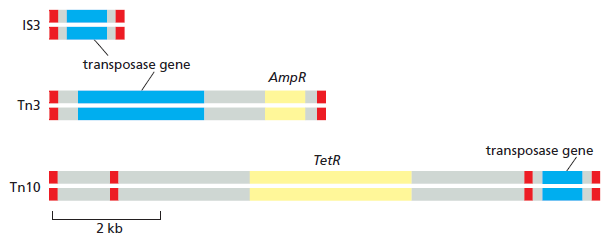
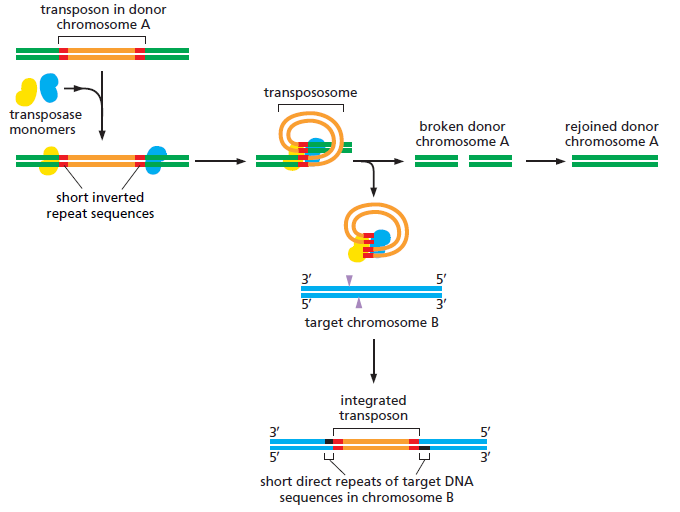
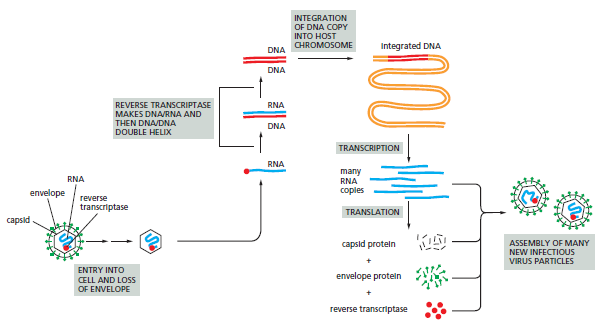

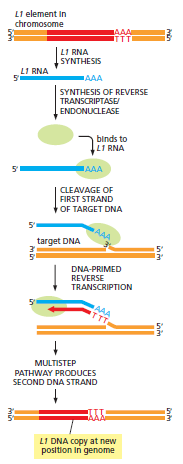


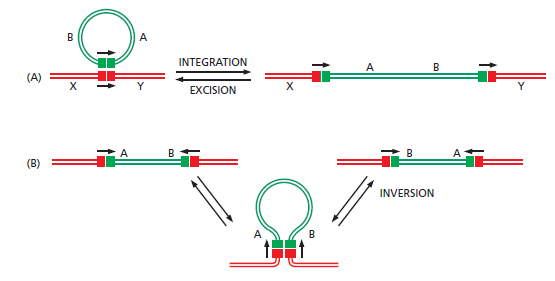
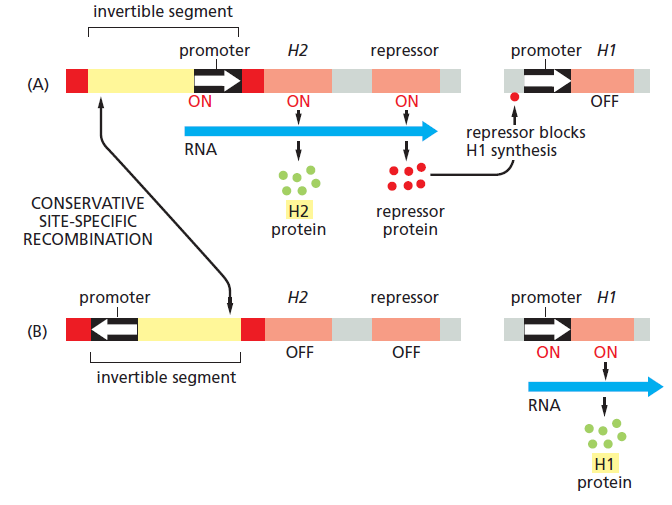
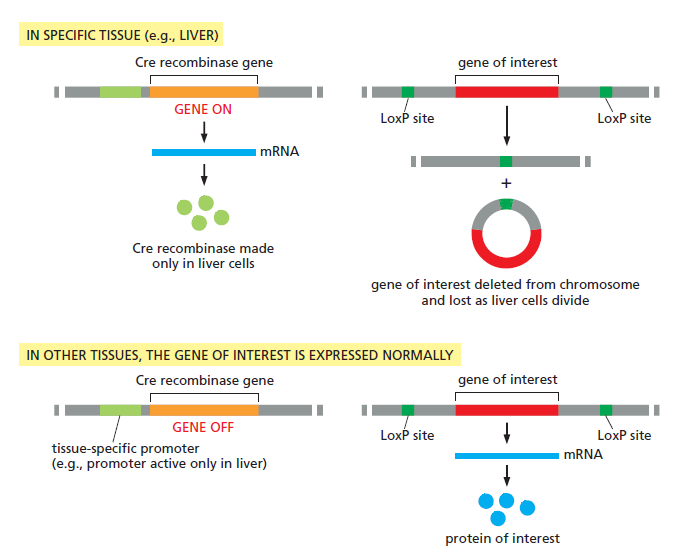
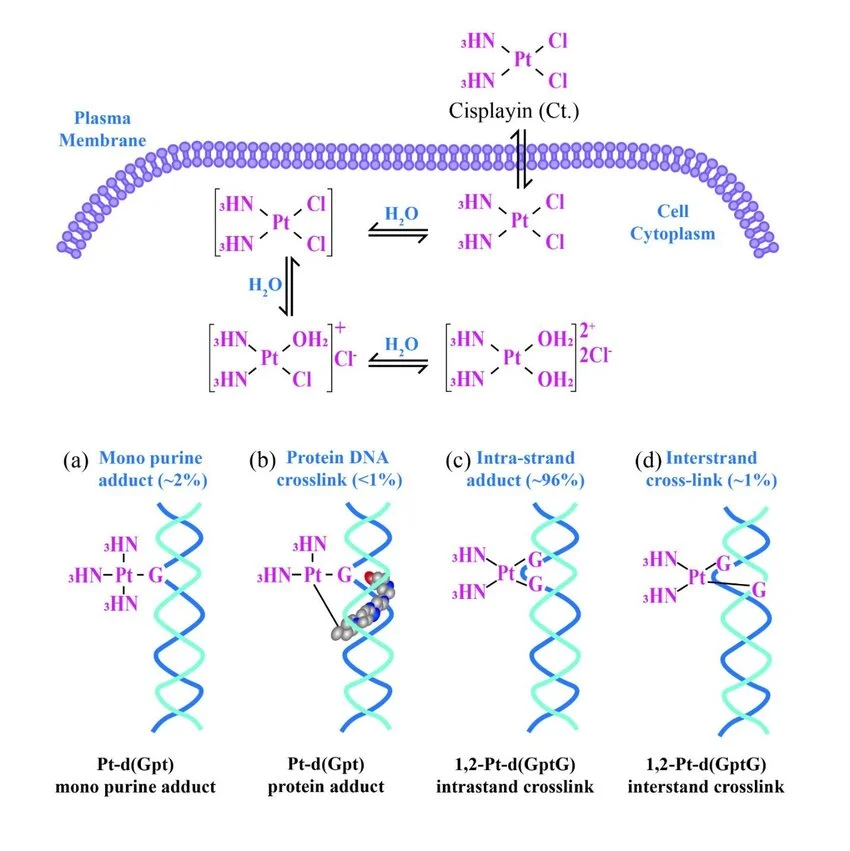
课堂问题:为什么NER缺陷(如着色性干皮病)患者既对铂类高度敏感,同时又容易因未修复损伤发展为癌症?
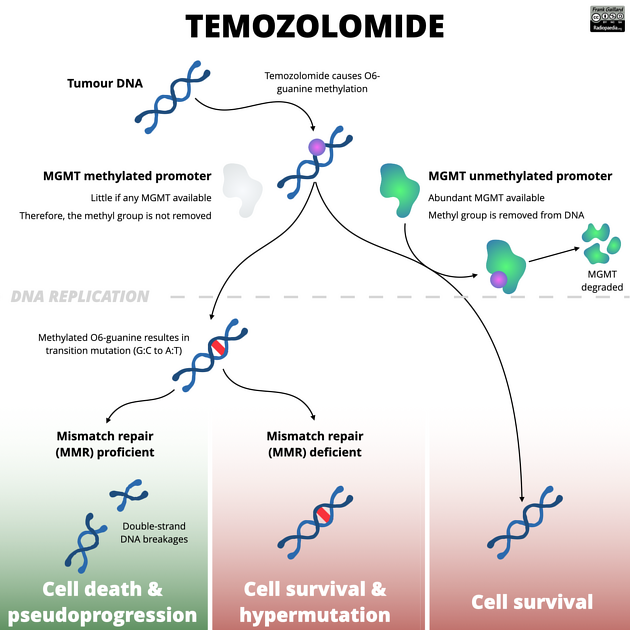
课堂问题:若肿瘤细胞MGMT启动子未甲基化,临床上应如何调整治疗策略?
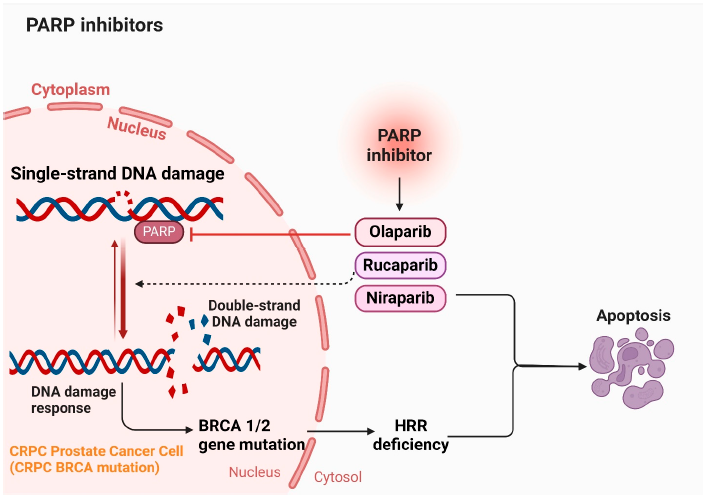
课堂问题:合成致死策略在没有可靠生物标志物筛选患者的情况下,会面临哪些临床挑战?
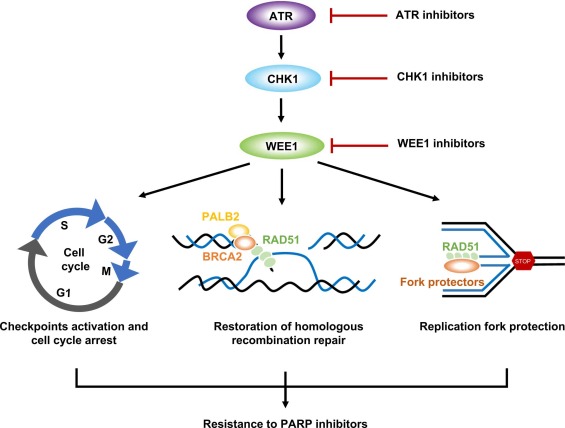
课堂问题:为什么检查点抑制剂通常不作为单药使用,而需要与DNA损伤剂联合?
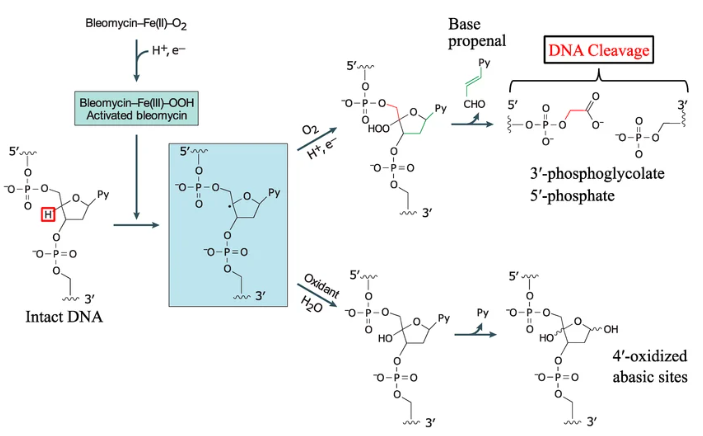
课堂问题:博来霉素所致的肺纤维化毒性与肺细胞的DNA修复能力有何关联?
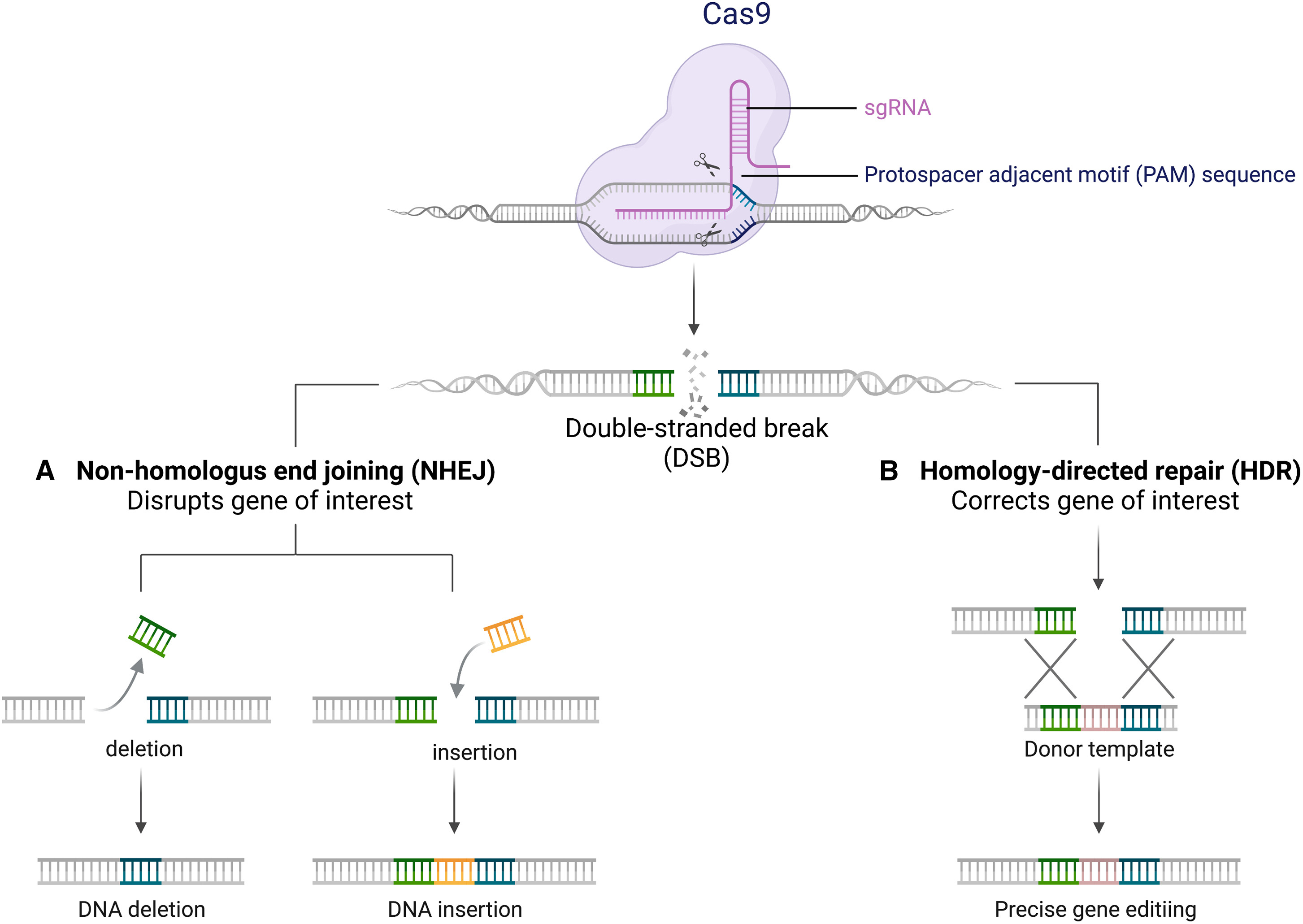
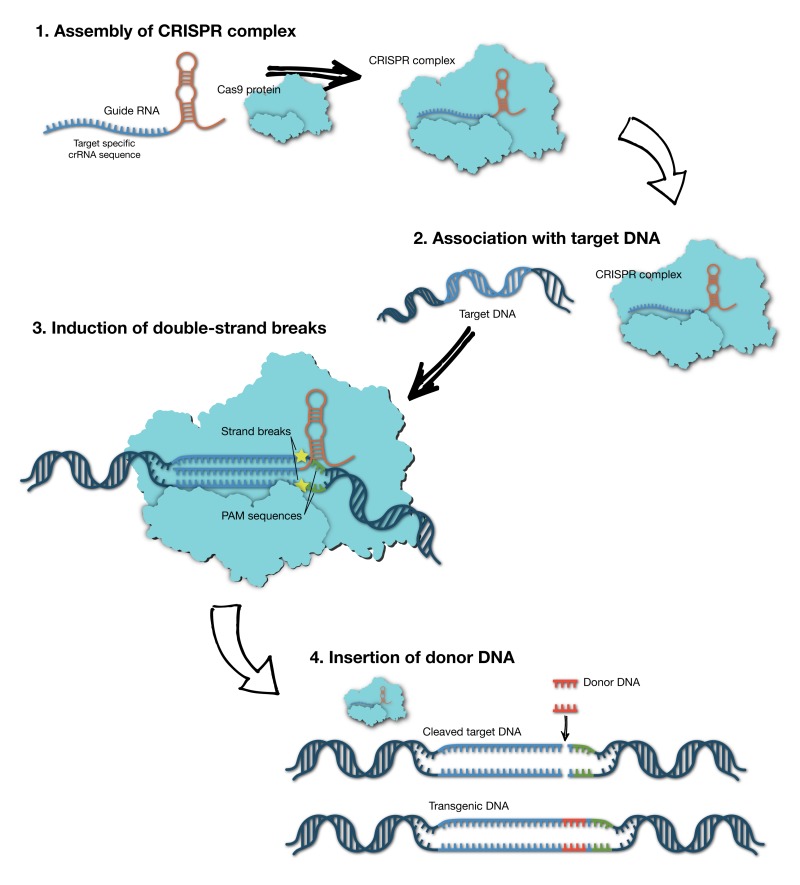
课堂问题:CRISPR编辑效率高度依赖HDR还是NHEJ修复通路——这与细胞周期和细胞类型有何关联?

课堂问题:溶瘤腺病毒ONYX-015在p53突变细胞中选择性复制的分子基础是什么?为何临床结果不如预期?
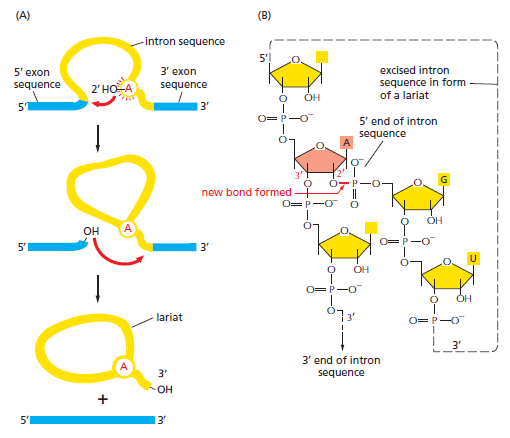
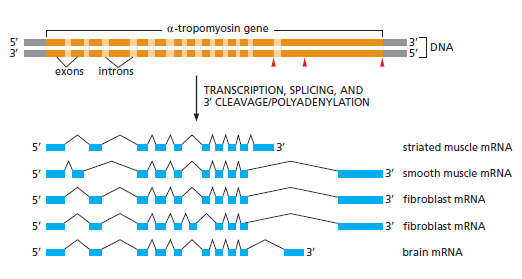
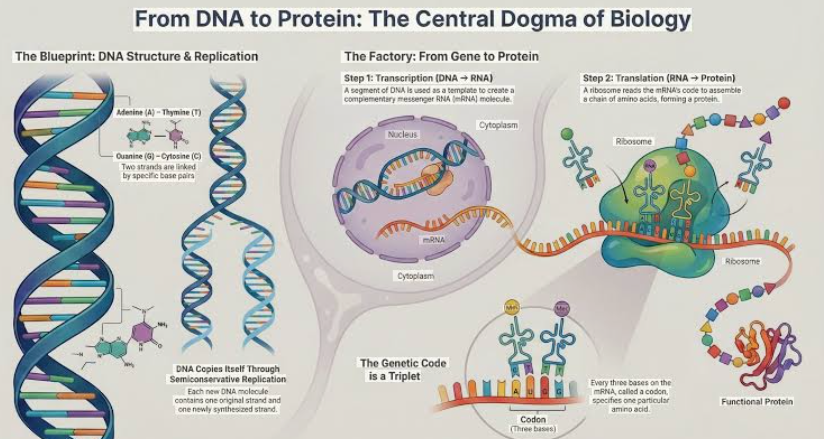
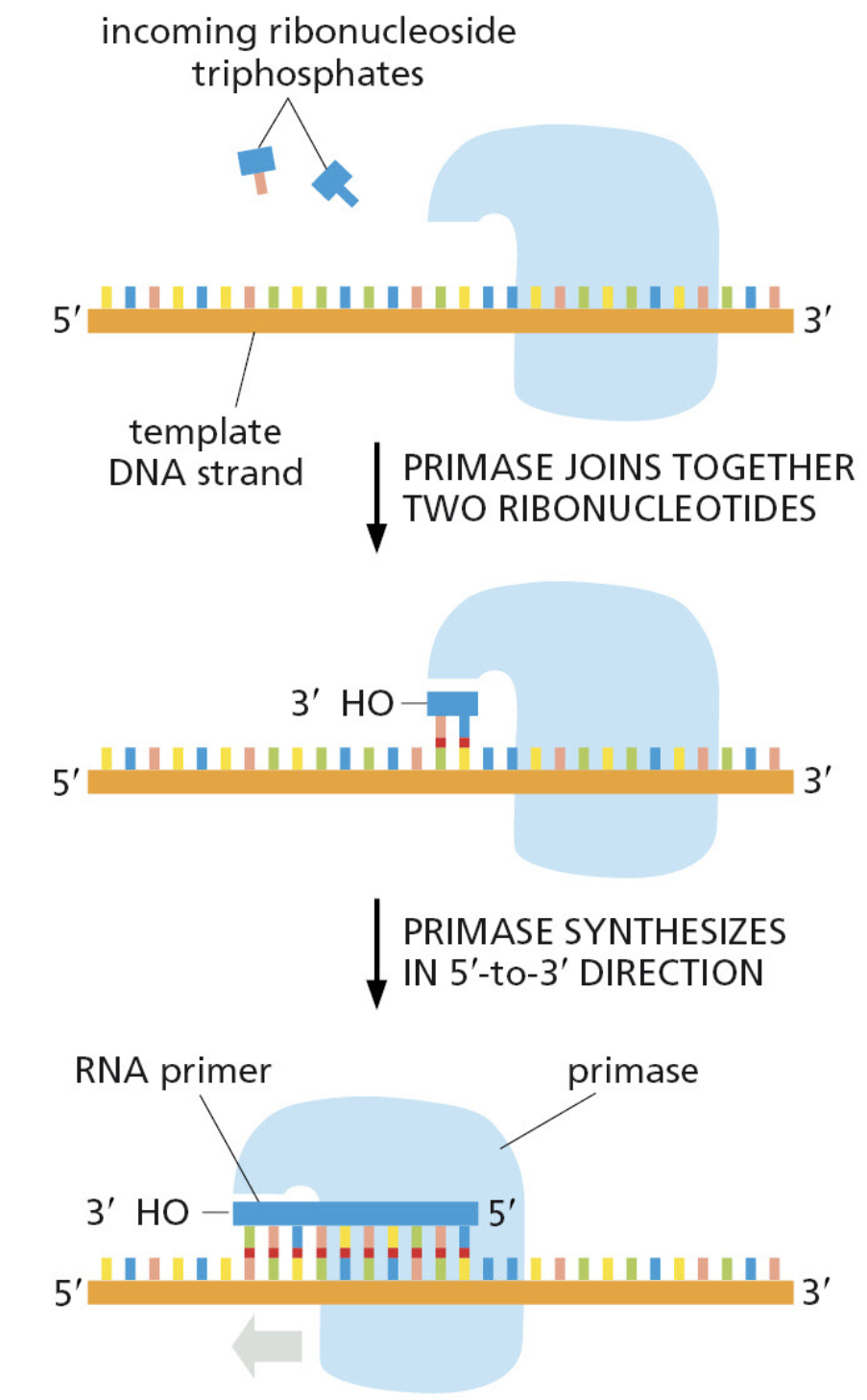
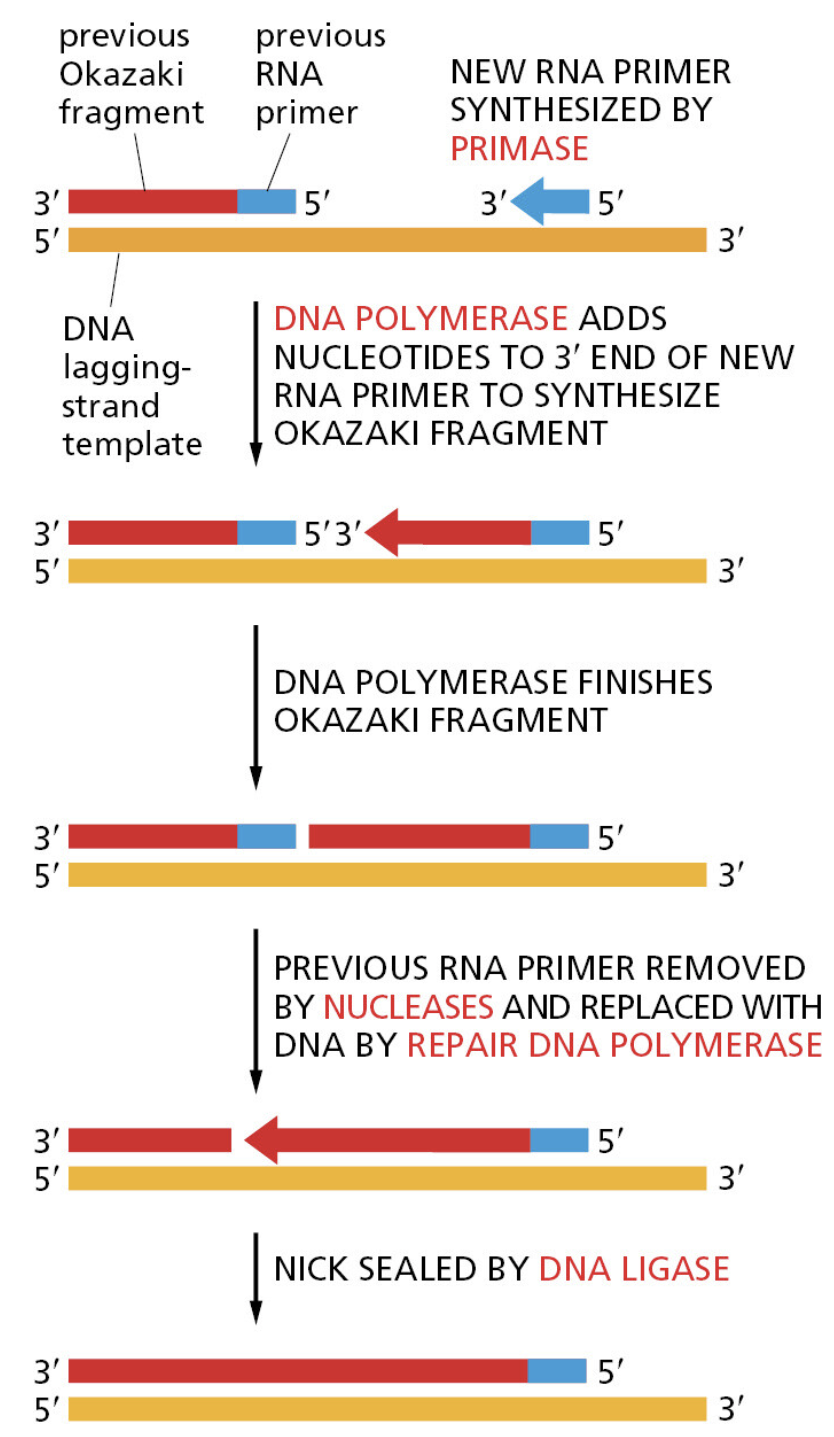
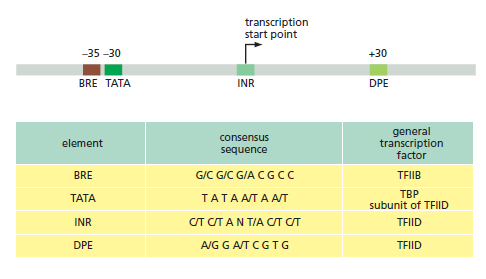
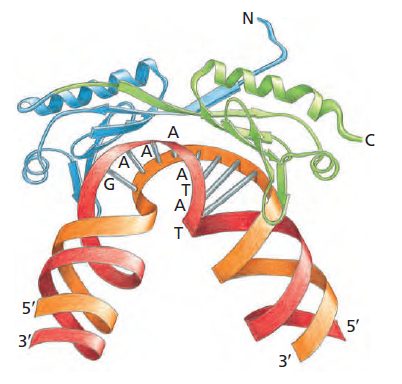
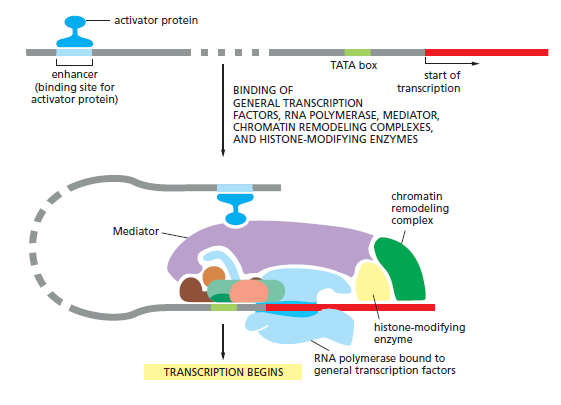
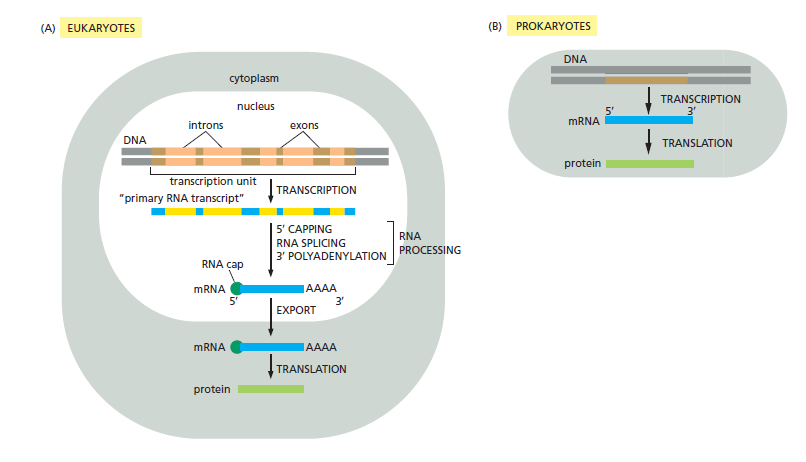
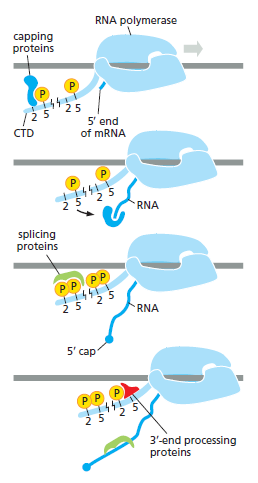
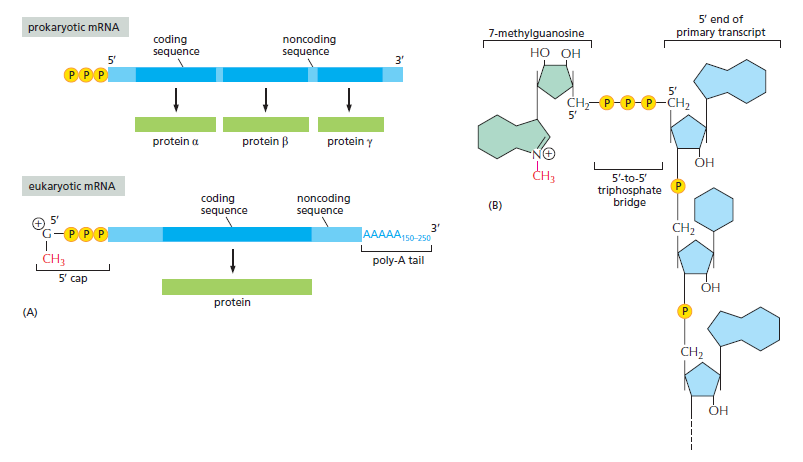
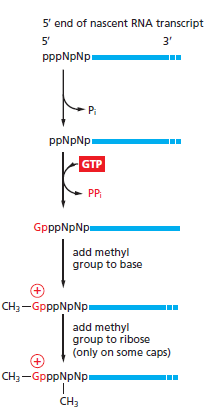
本讲核心问题:细胞如何精确、可控、可调节地把 DNA 转换成 RNA?为什么真核细胞的转录如此复杂、还需要那么多加工步骤?
教学要点:"基因表达"远不止"做蛋白质"—— 大量 RNA 本身就是终产物,是细胞调控网络的核心。理解转录机制 = 理解整个细胞功能的"开关系统"。
从这里出发:本讲的每一个机制(启动子识别、剪接、polyA、表观调控)都对应着至少一类已上市的药物或正在研发的疗法 —— 理解转录 = 理解 21 世纪药学创新的核心引擎。
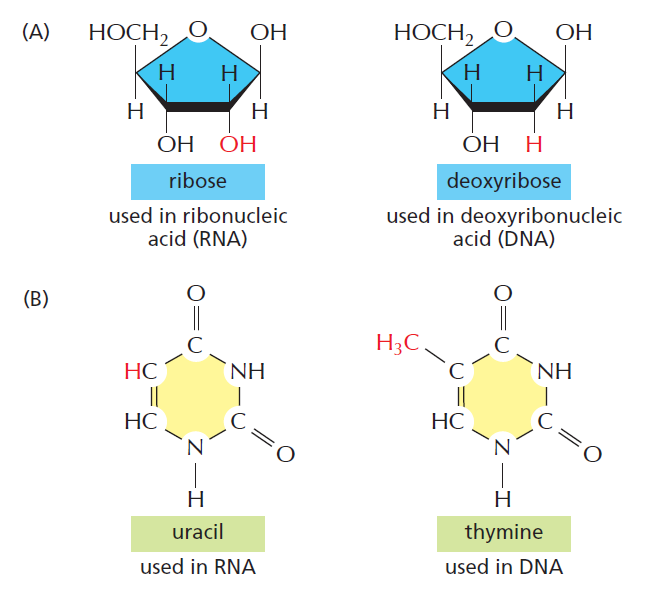
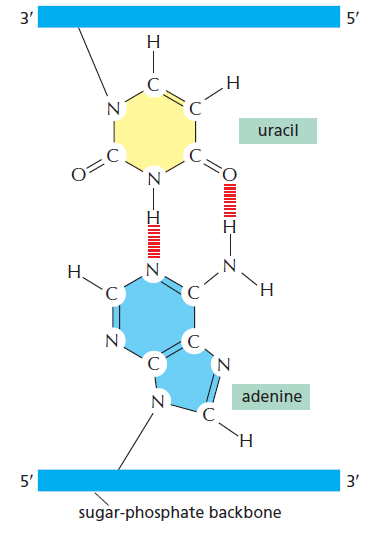
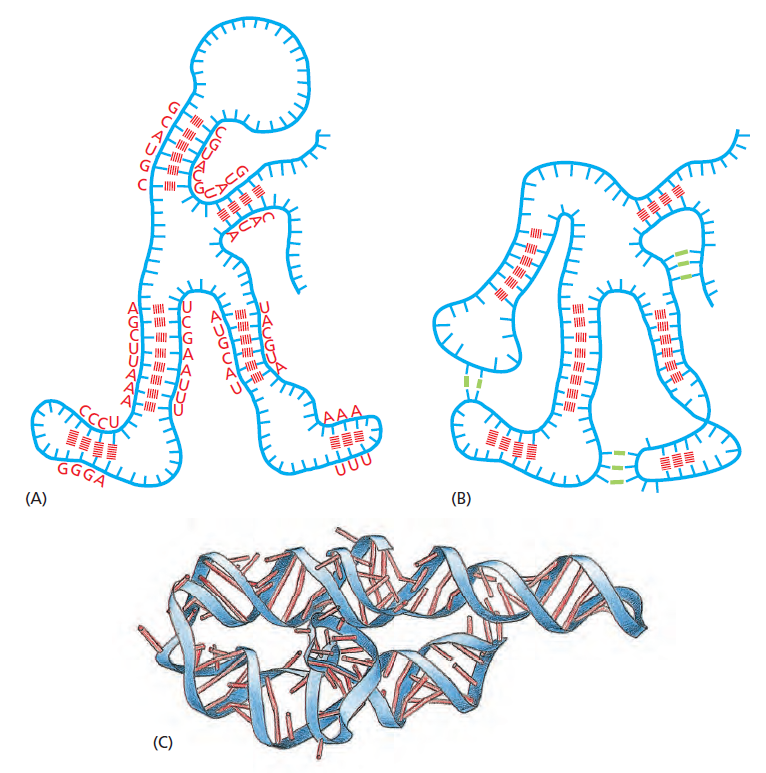
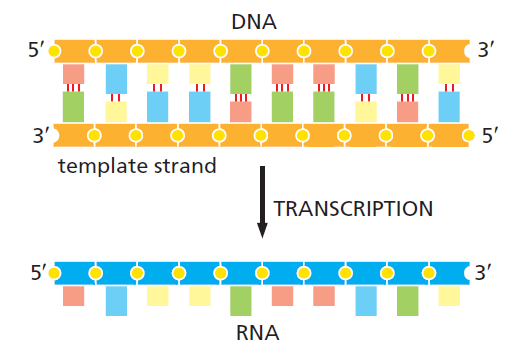
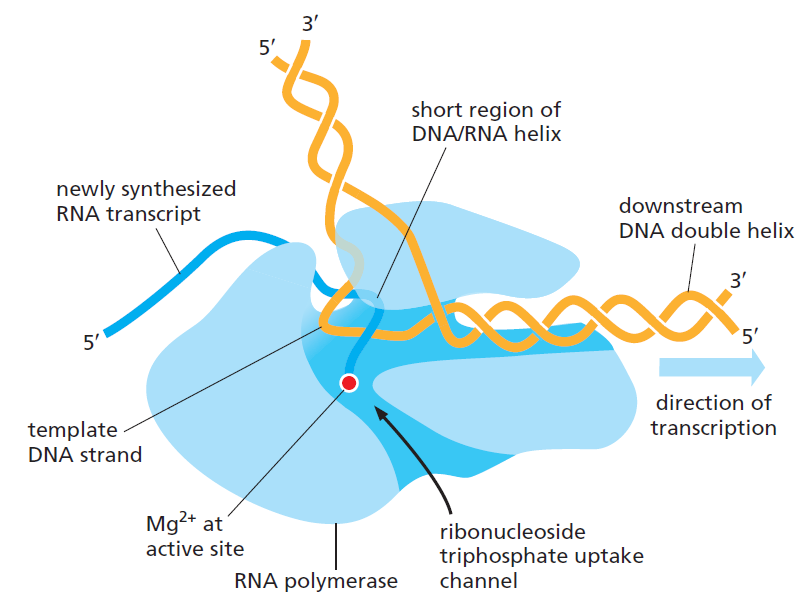
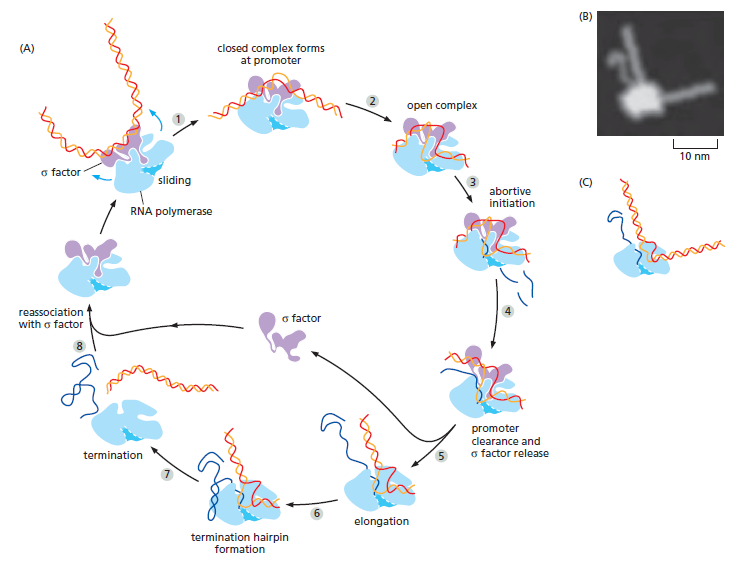
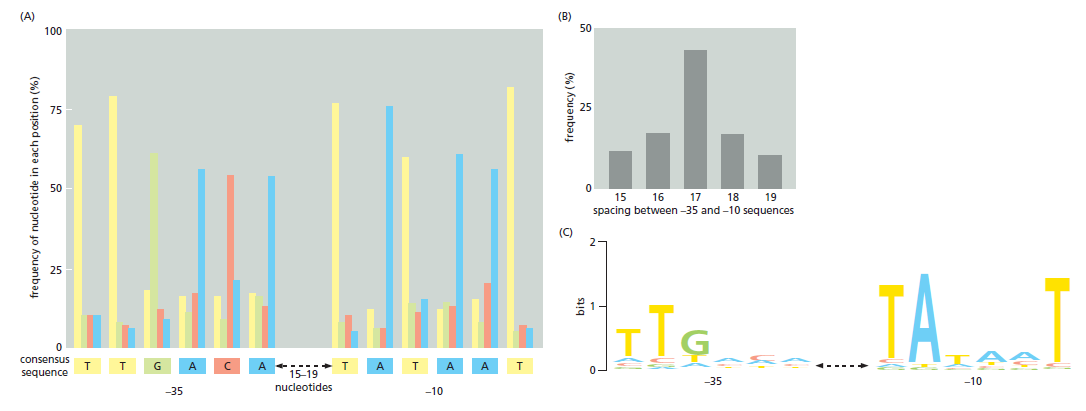
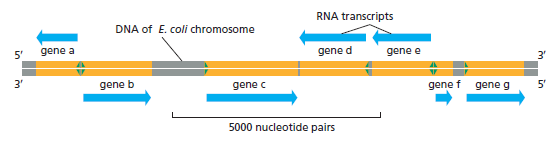
| 聚合酶 | 转录基因类型 |
|---|---|
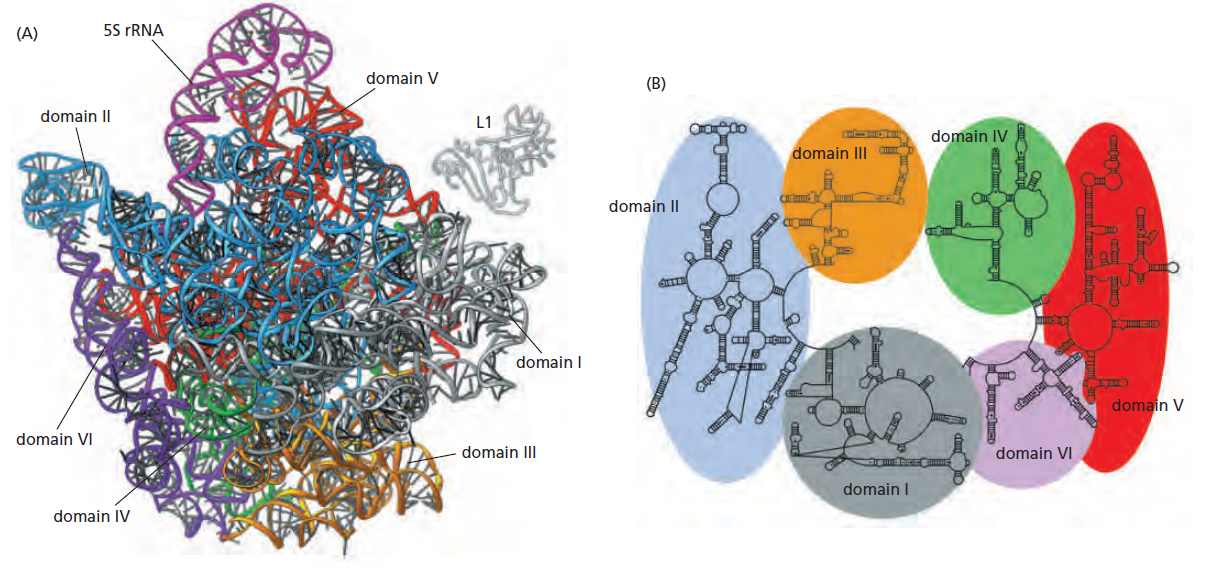
| RNA Pol I | 5.8S, 18S, 28S rRNA |
| RNA Pol II | 蛋白编码基因 + 多种ncRNA |
| RNA Pol III | tRNA, 5S rRNA, 部分snRNA |
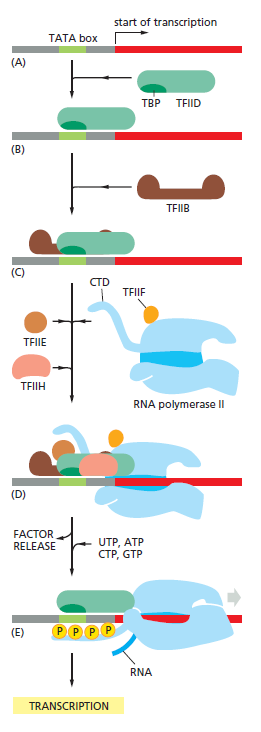
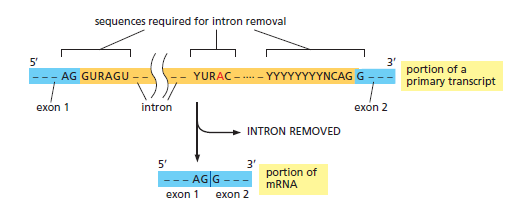
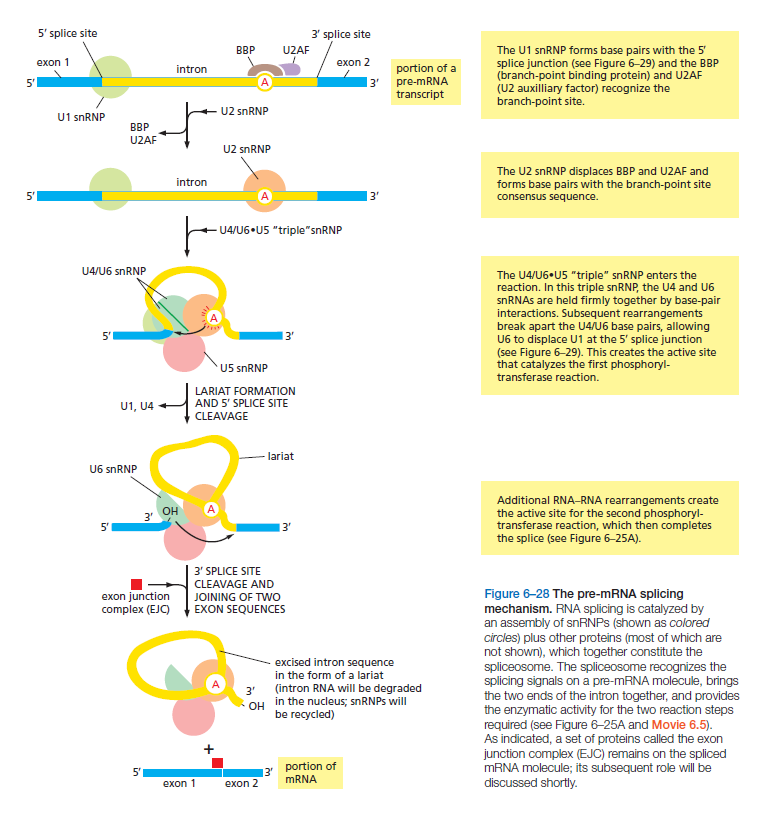
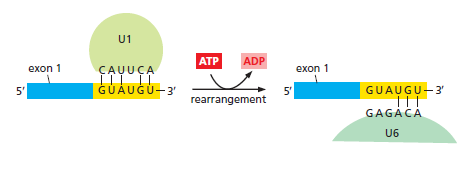
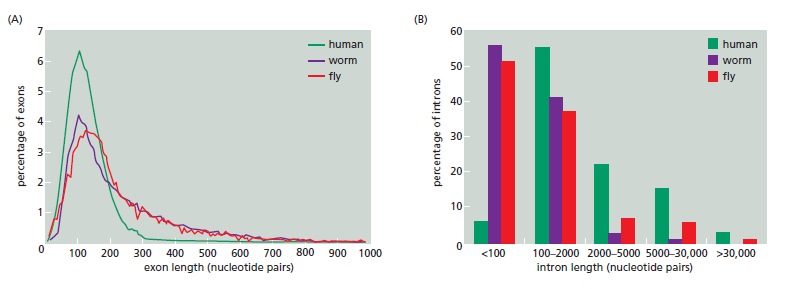
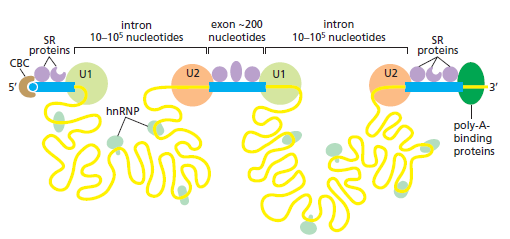
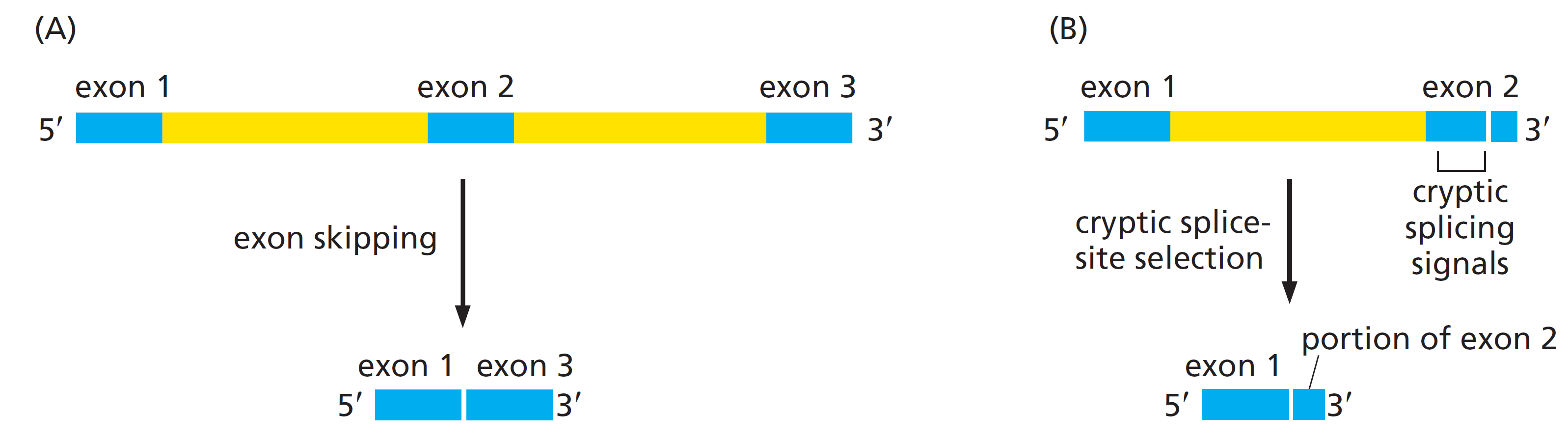
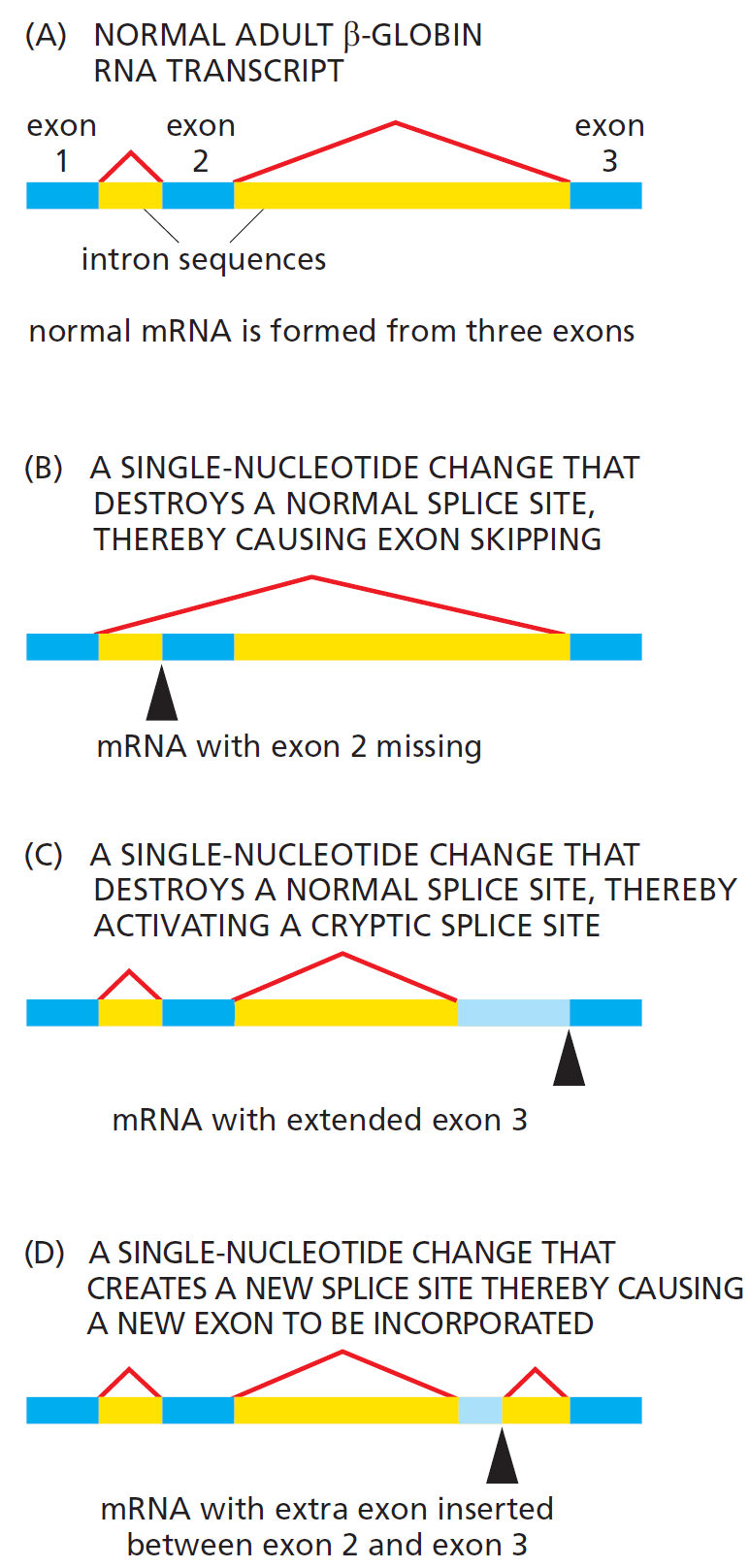
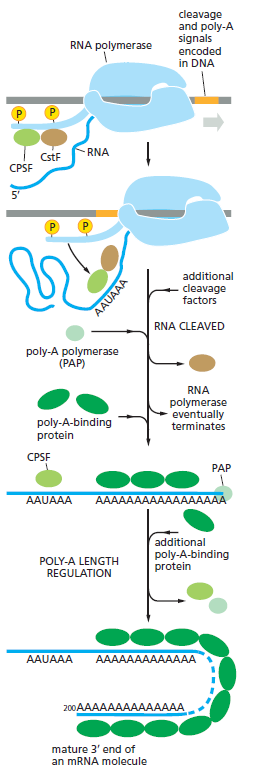
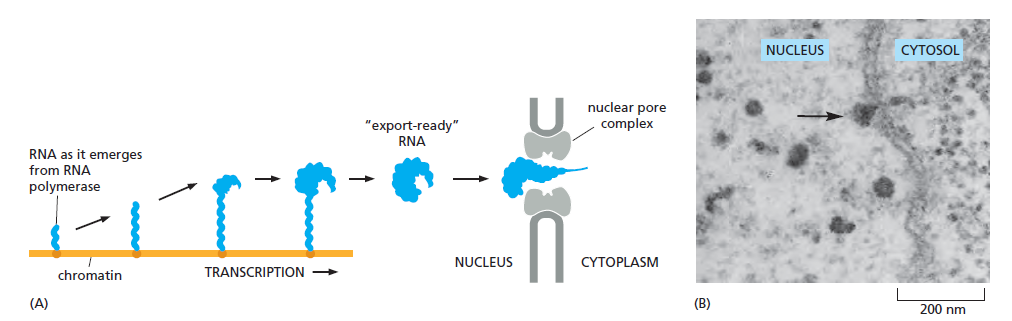
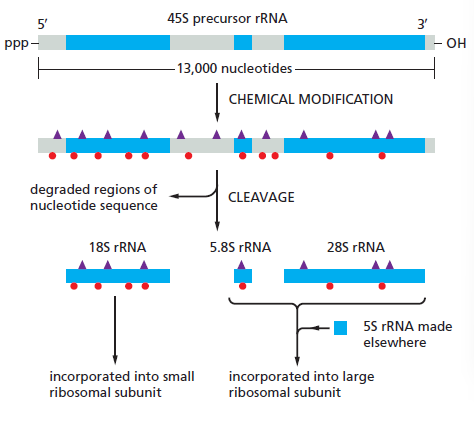
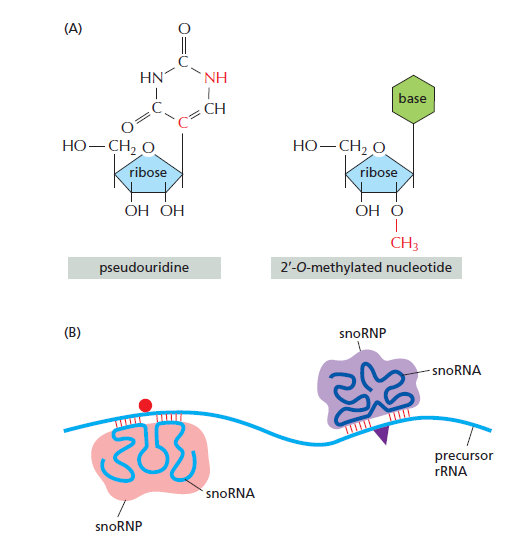
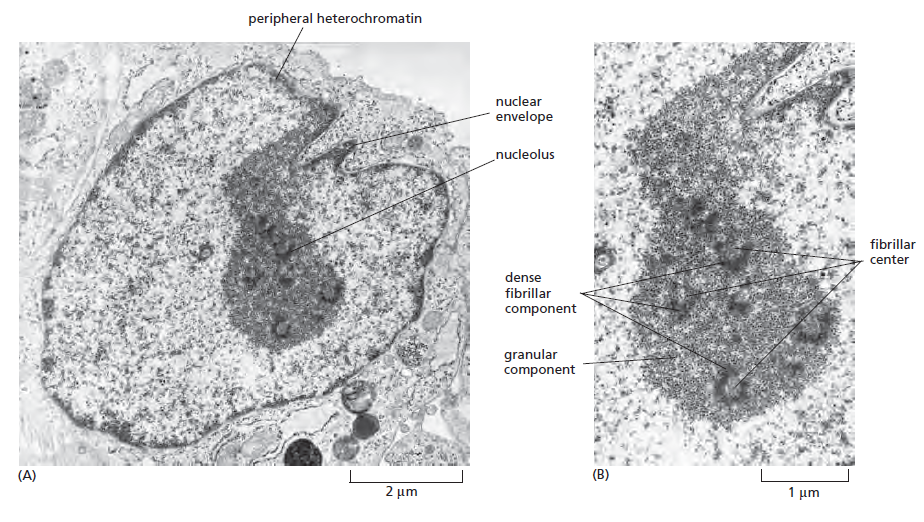
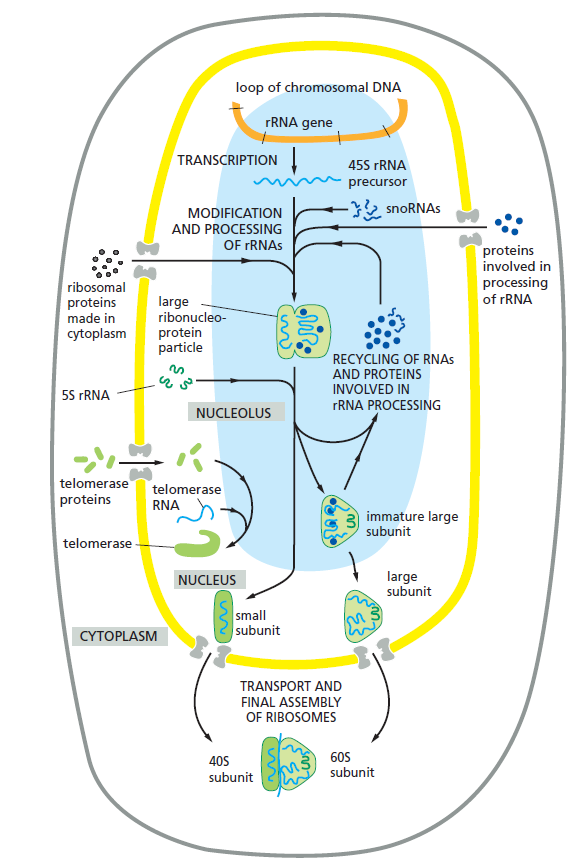
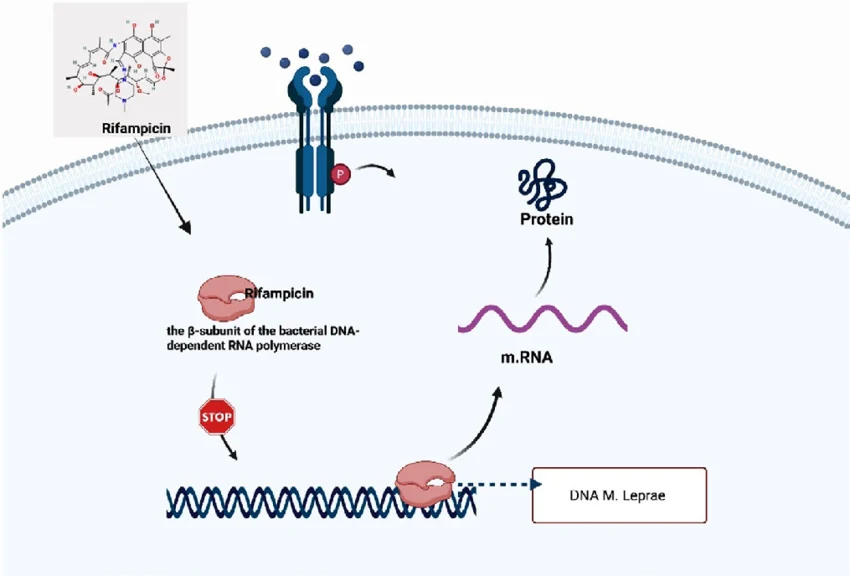
课堂问题:结核分枝杆菌中rpoB基因突变是最常见的利福平耐药机制,这对联合用药策略有何启示?
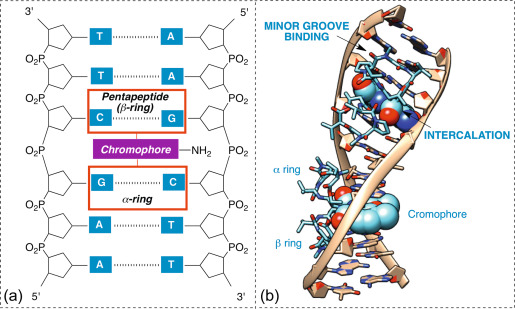
课堂问题:放线菌素D既抑制转录又抑制复制,在研究中如何利用这一特性区分两种过程的贡献?
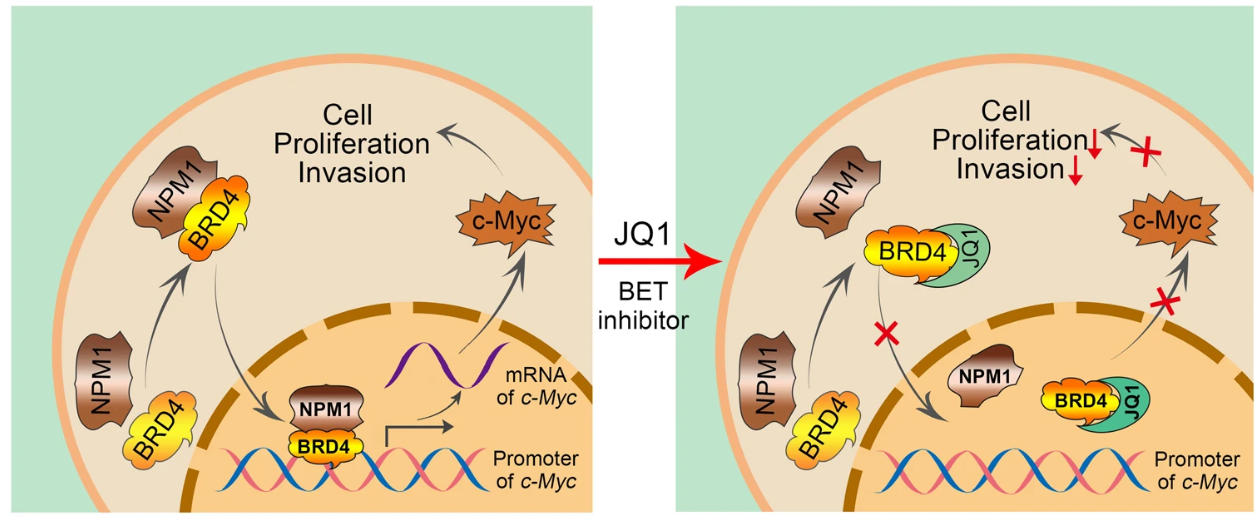
课堂问题:MYC本身难以直接成药,BET抑制剂如何"间接靶向"MYC?这一策略有何局限?
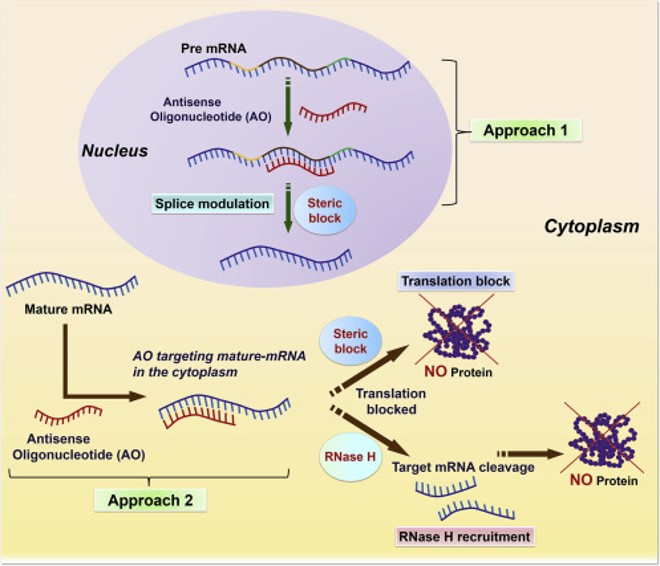
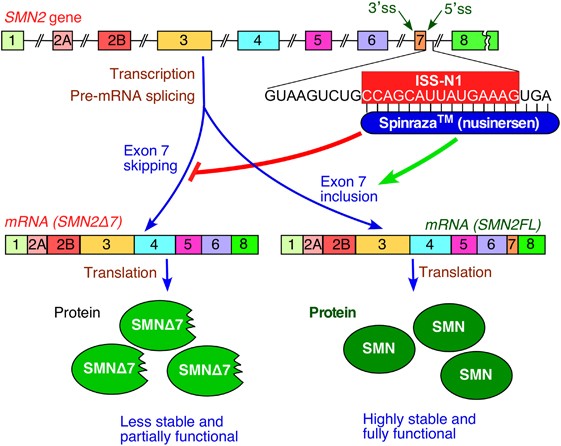
课堂问题:同样靶向SMN2剪接,口服小分子Risdiplam与鞘内ASO Nusinersen在机制和患者依从性上有哪些差异?
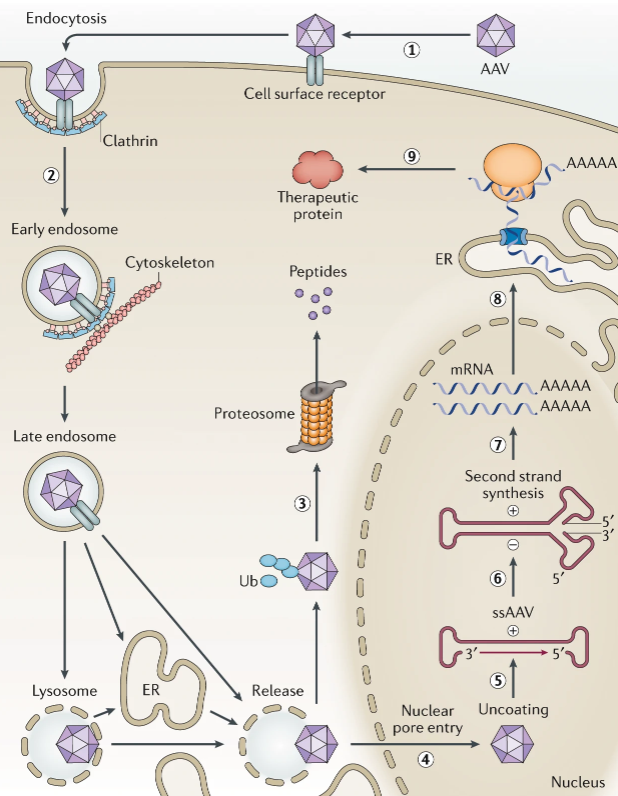
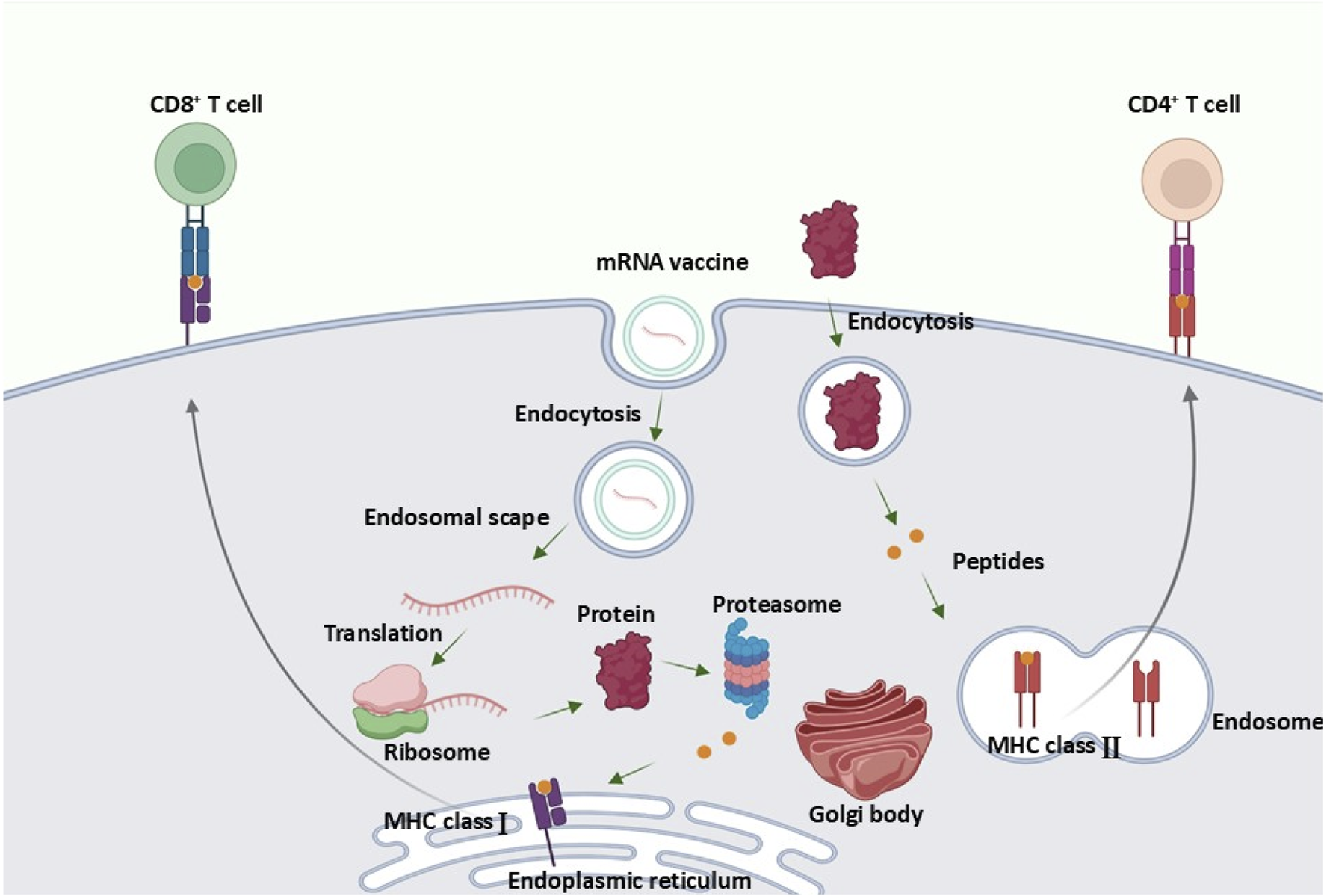
课堂问题:AAV疗法需要设计靶细胞特异性启动子,mRNA疗法则需要优化5'UTR与密码子——两者分别利用了转录与翻译调控的哪些原理?
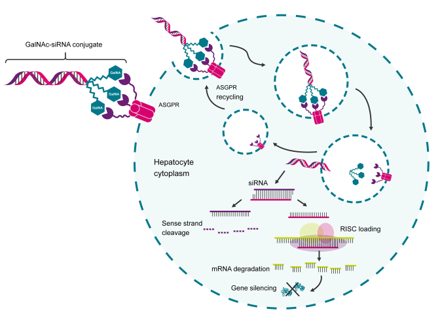
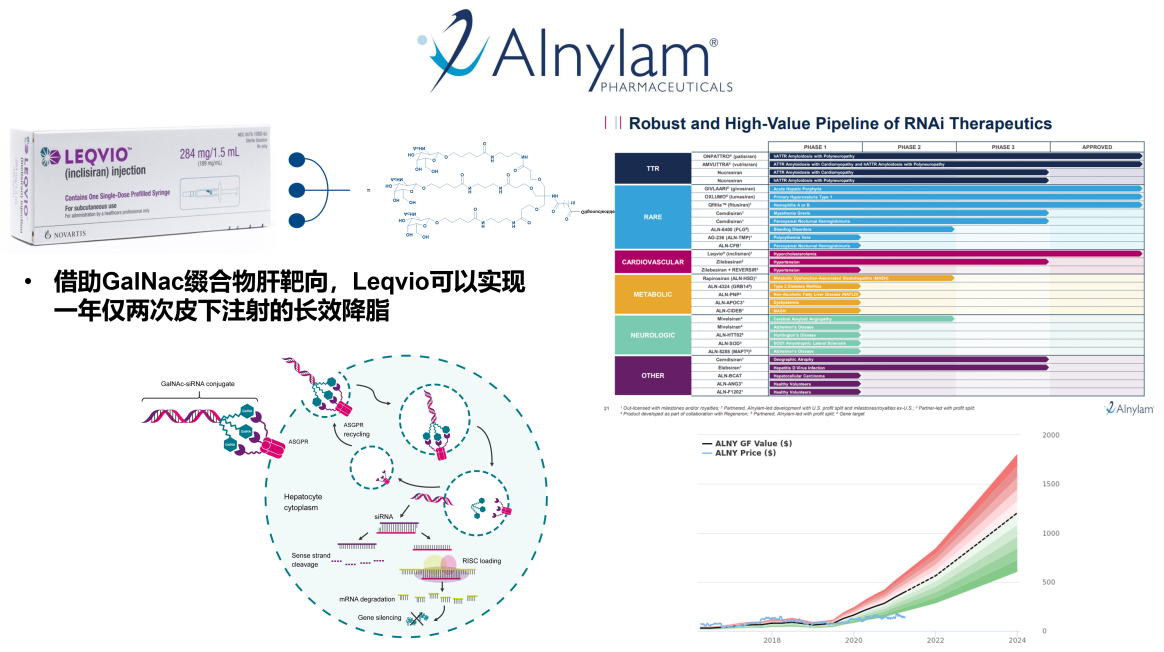
课堂问题:GalNAc修饰的siRNA(Inclisiran)为何可以实现每年仅需注射2次的超长效维持?与LNP递送相比有何优劣?
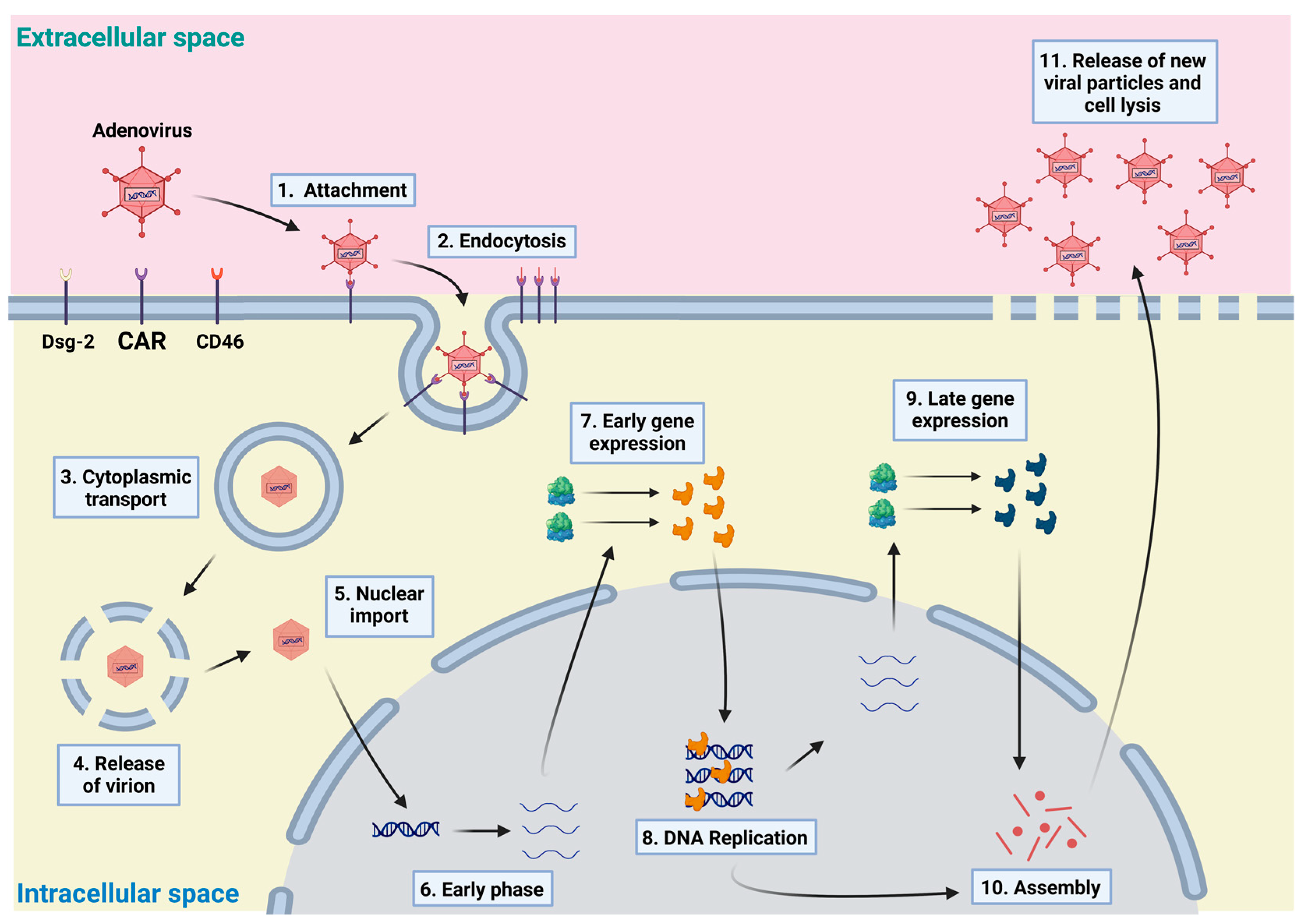
课堂问题:腺病毒载体不整合宿主基因组,目的基因的转录是否会持久?与AAV和逆转录病毒载体相比有何取舍?
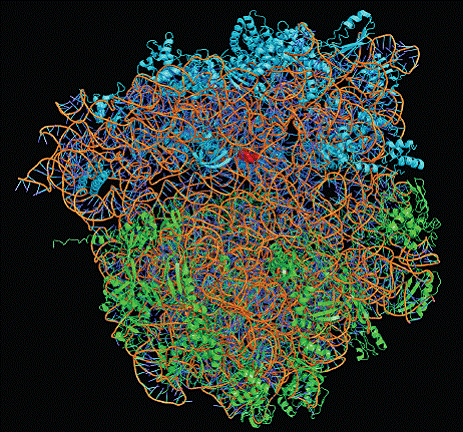
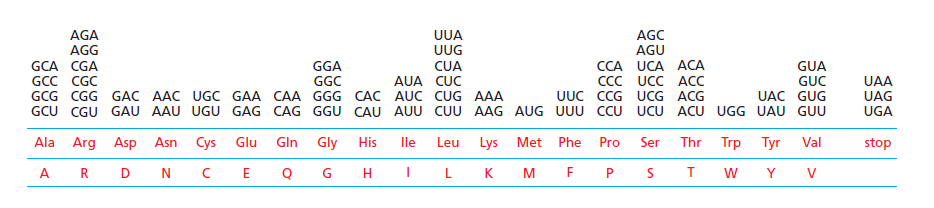
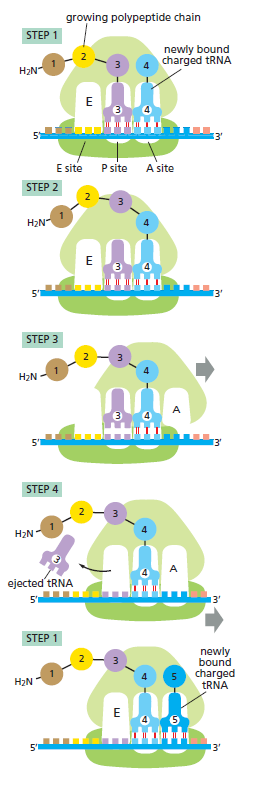
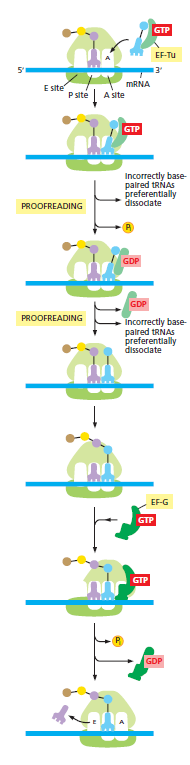
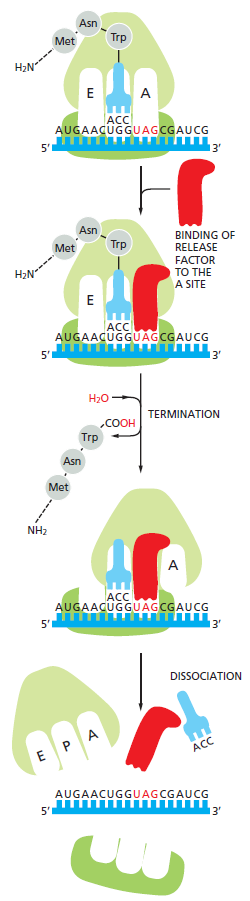
本讲核心问题:核糖体如何在三种不同 RNA(mRNA + tRNA + rRNA)的协同下,把核苷酸语言精确翻译成蛋白质语言?错译会带来什么后果?
本讲学习目标:不仅要理解"如何翻译"的机制,更要看到核糖体这个"古老分子机器"如何成为抗生素、化疗、罕见病治疗的关键靶点 —— 翻译机制是连接基础研究与药物开发的重要纽带。

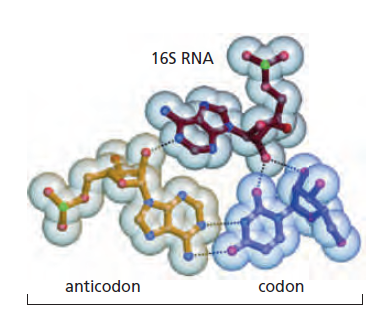
UUC-GAC-AUG-... 偏移 1 位变为 UCG-ACA-UGN-...,氨基酸完全不同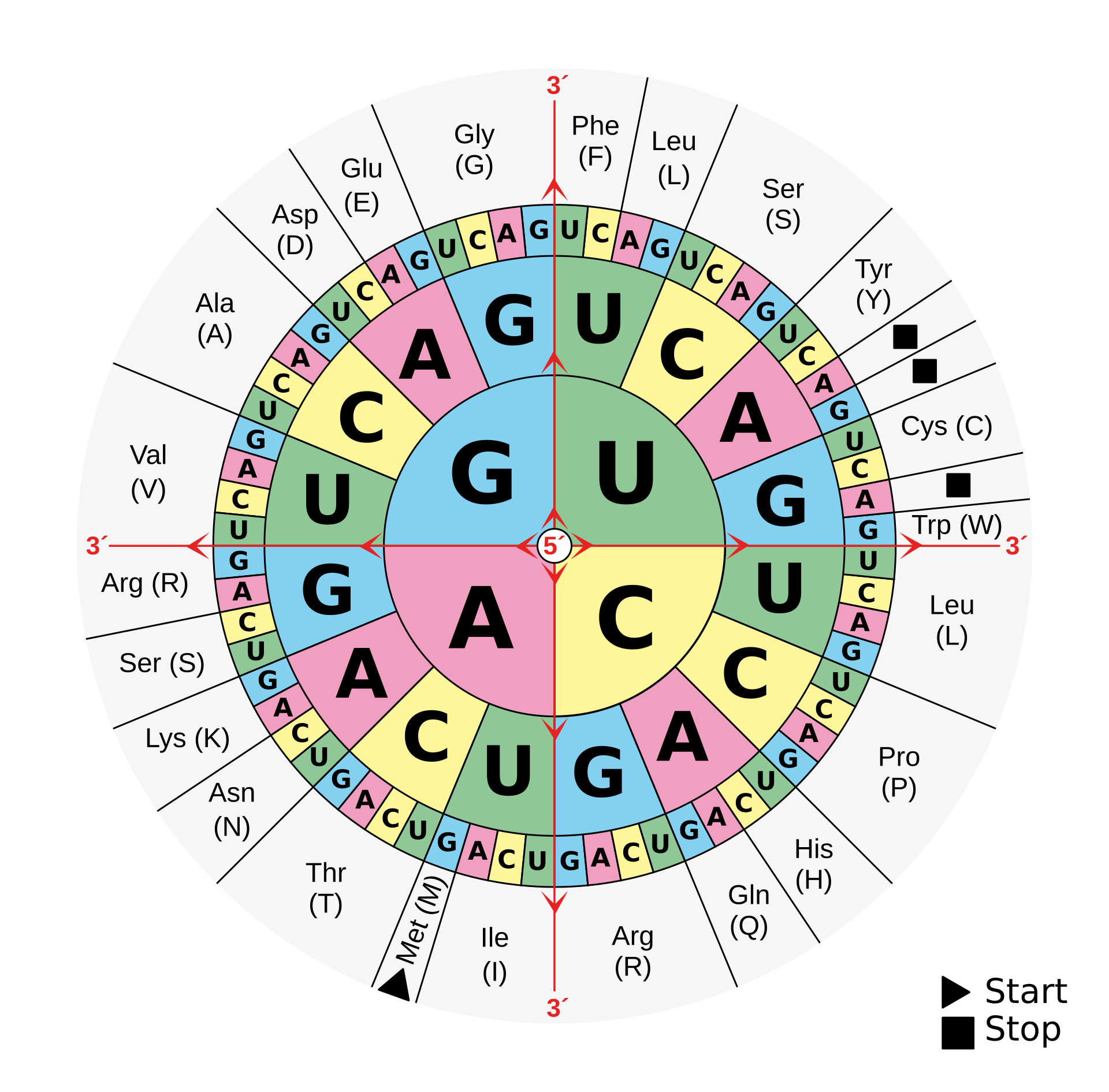
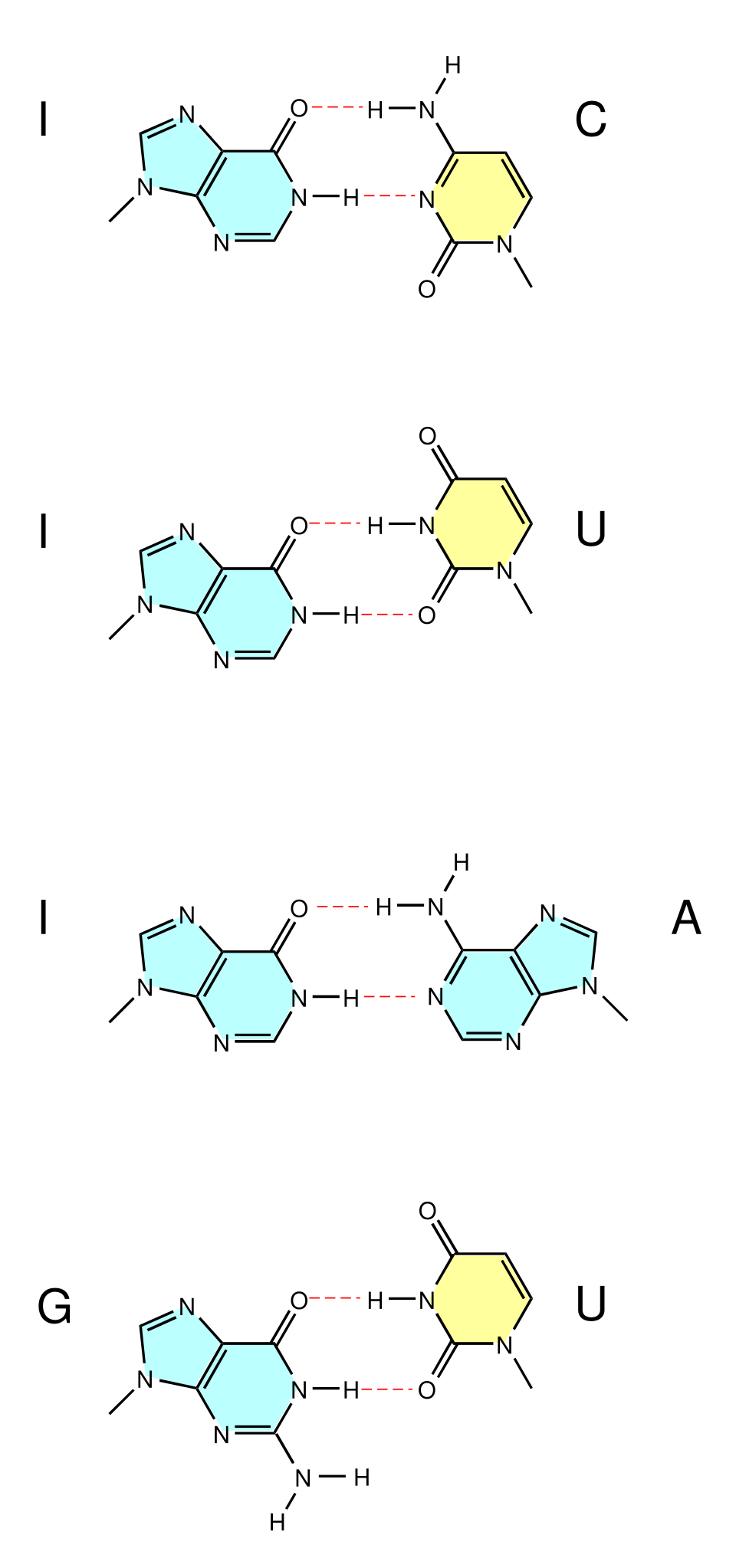
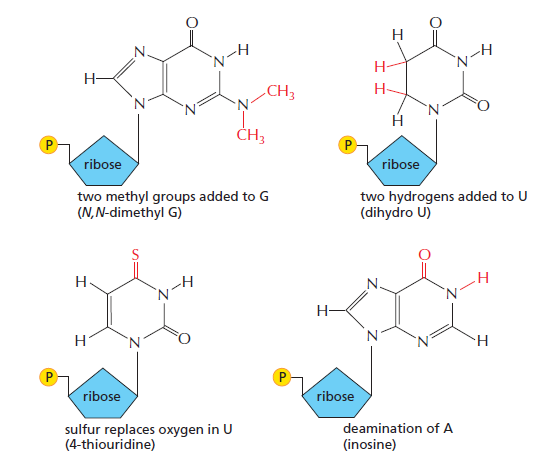
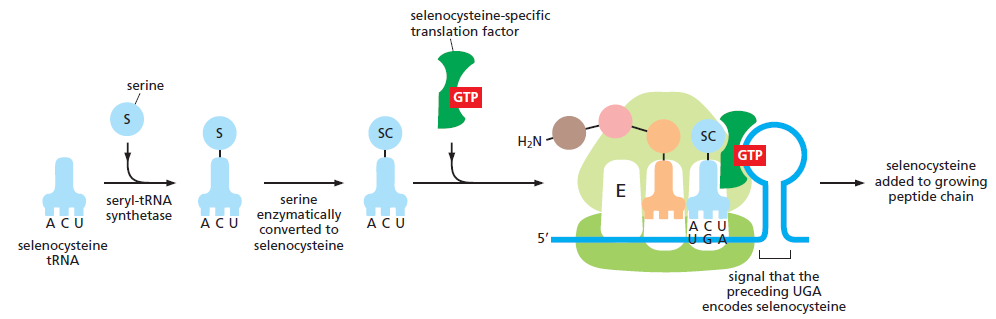
I(次黄嘌呤 inosine)→ 可同时配 U / C / A;若为 G → 可配 U / C;若为 U → 可配 A / G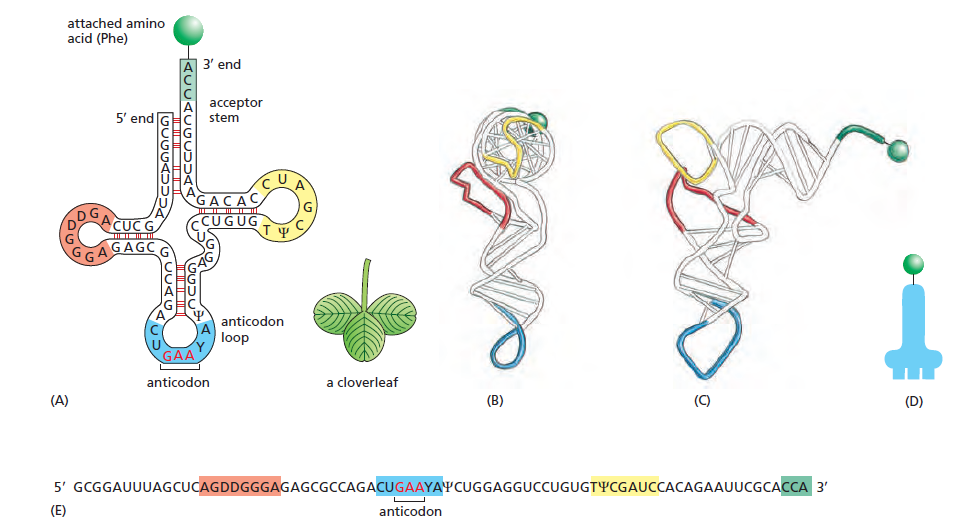
Cys-tRNACysAla-tRNACys(反密码子还认 Cys 密码子 UGU/UGC,但带的是 Ala)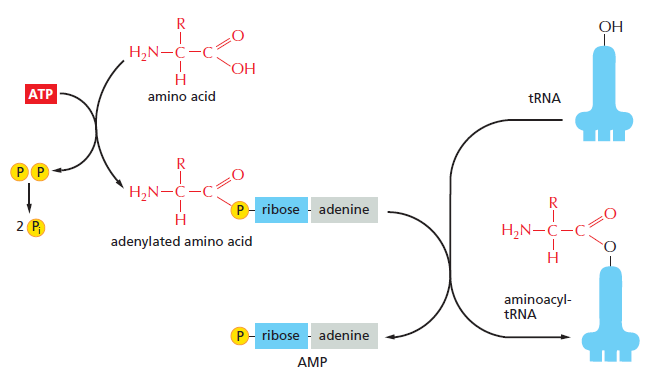
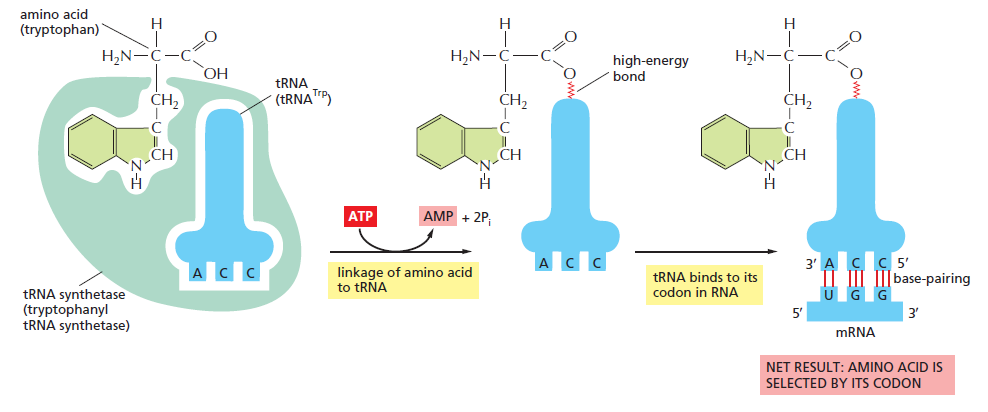
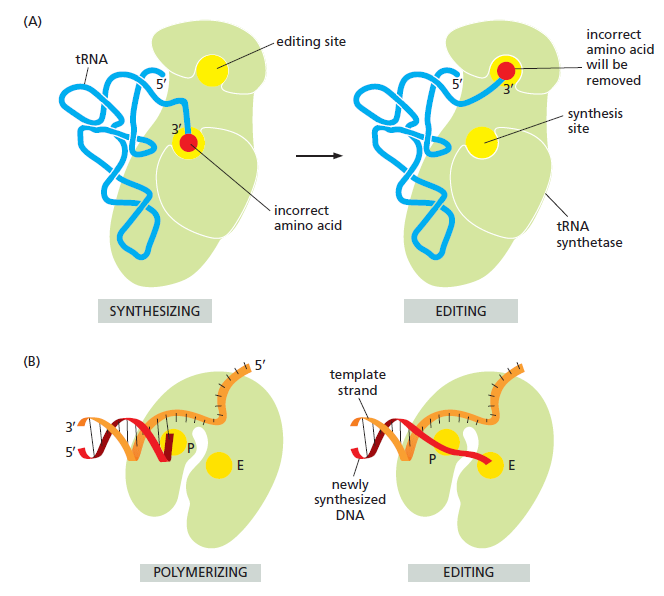
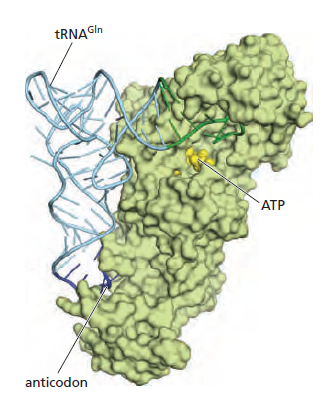
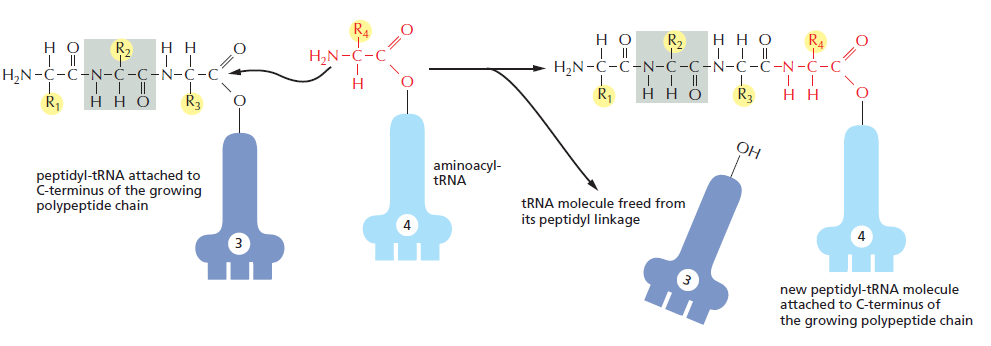
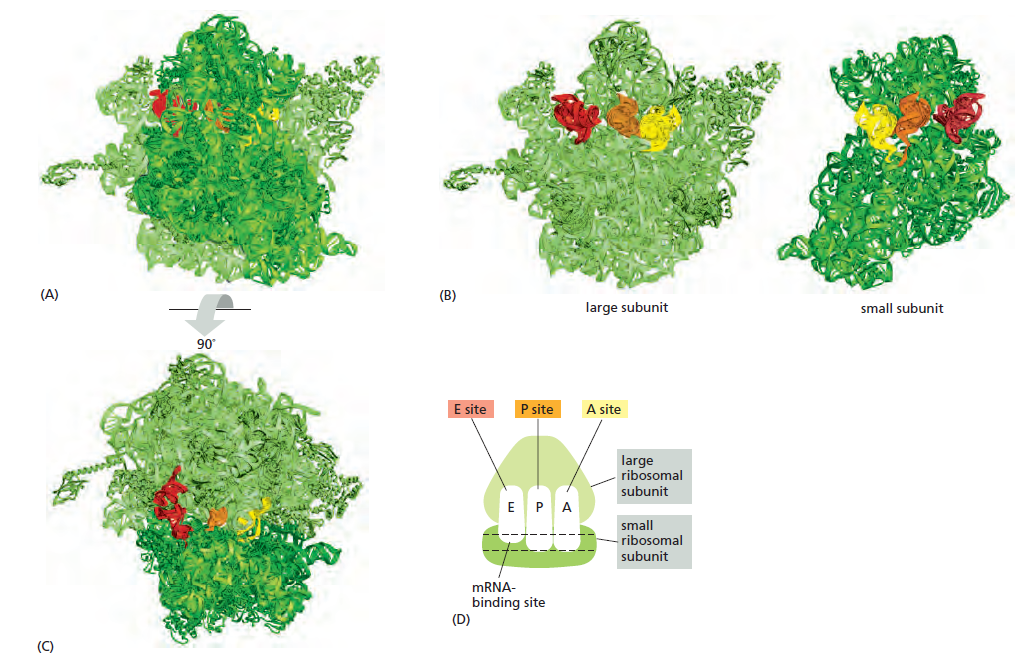

(gcc)gccRccAUGG(大写 = 高度保守关键位,小写 = 偏好位,R = 嘌呤 A/G)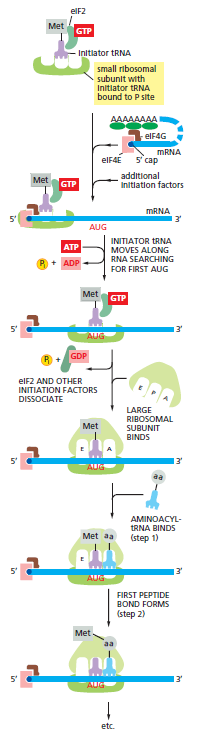
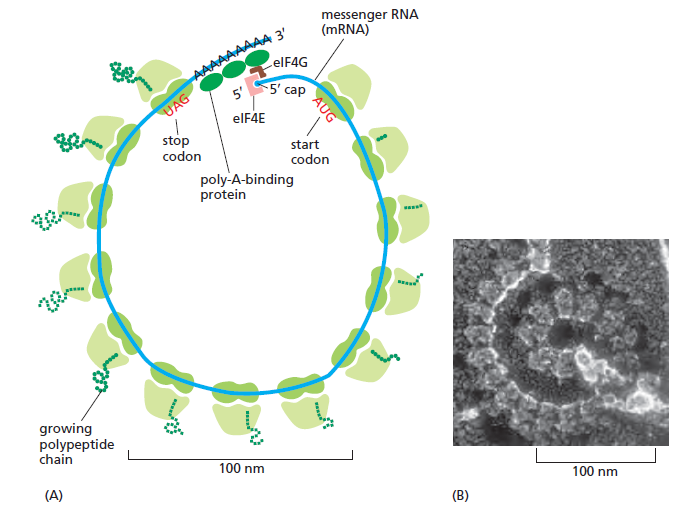
AGGAGG,位于起始 AUG 上游 ~7 nt)CCUCCU)碱基互补 → 把 30S 小亚基直接定位到 AUG,无需扫描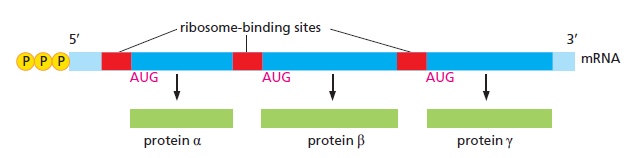
| 抗生素 | 作用靶点 |
|---|---|
| 四环素 | 阻止aminoacyl-tRNA结合A位 |
| 链霉素 | 阻止起始→延伸的转换,引起错读 |
| 氯霉素 | 阻断转肽酶反应 |
| 红霉素 | 结合出口通道,抑制肽链延伸 |
| 嘌呤霉素 | 模拟aminoacyl-tRNA → 引起提前终止 |
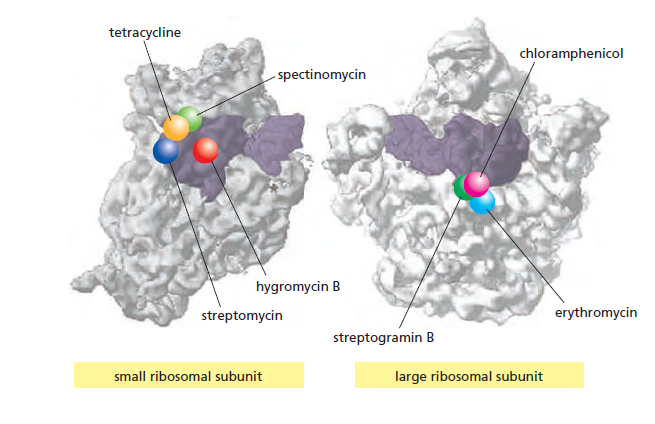

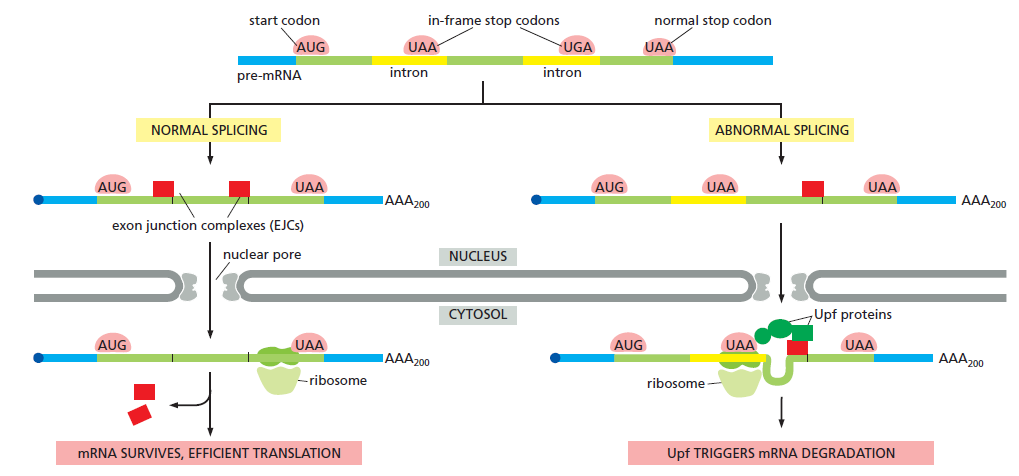
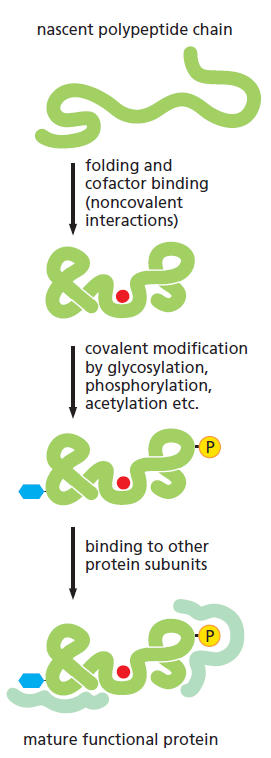
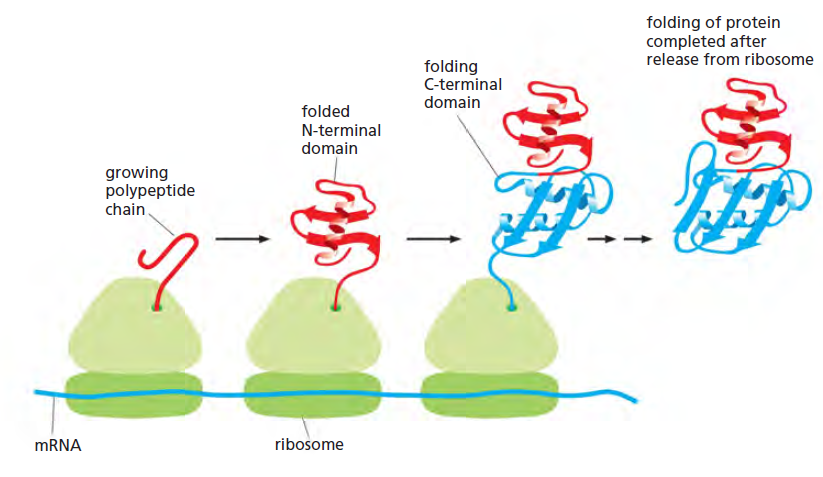
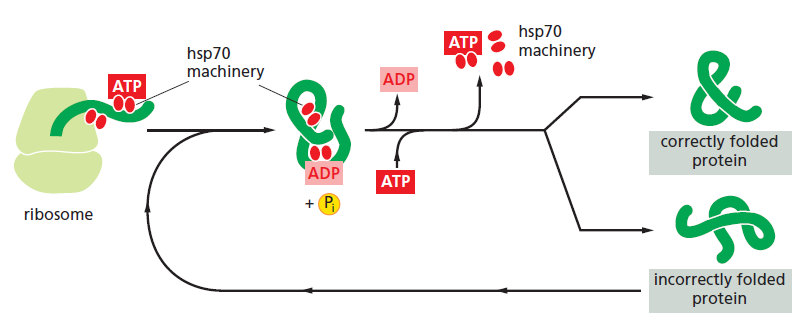
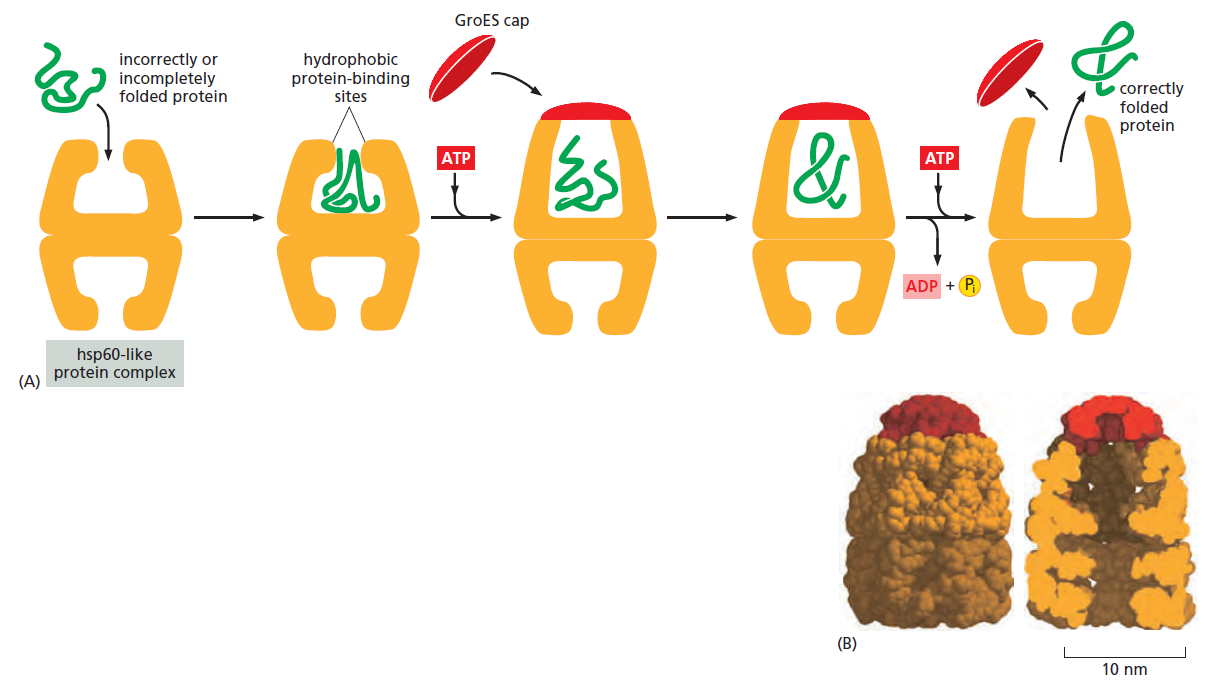
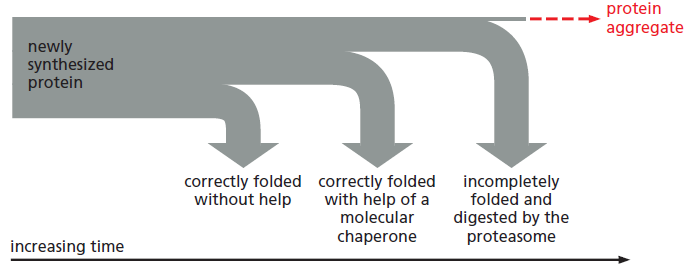
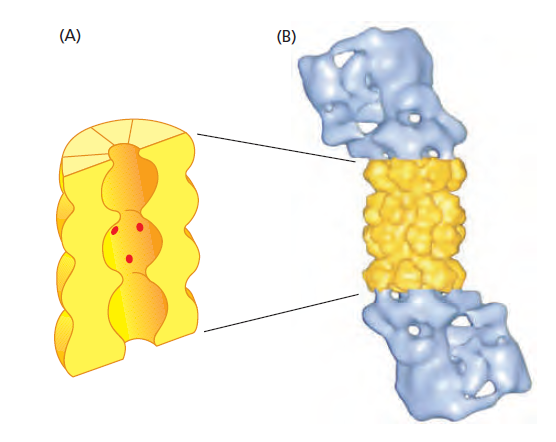
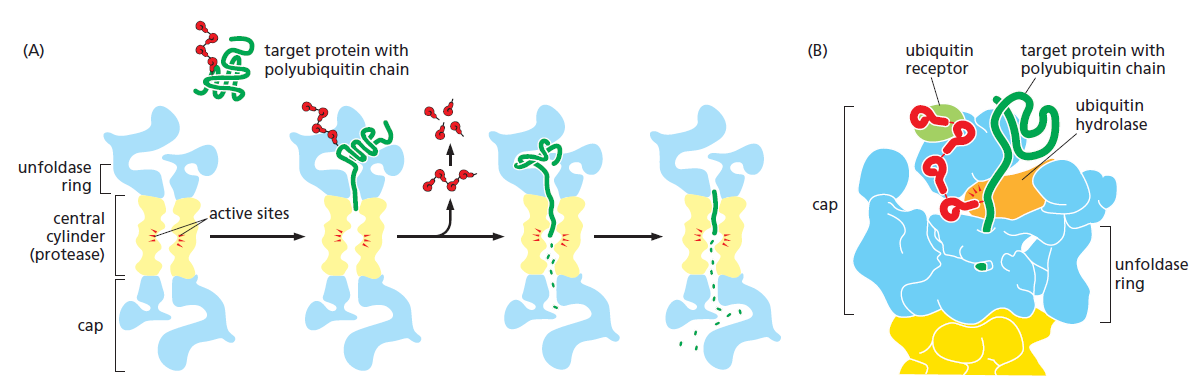
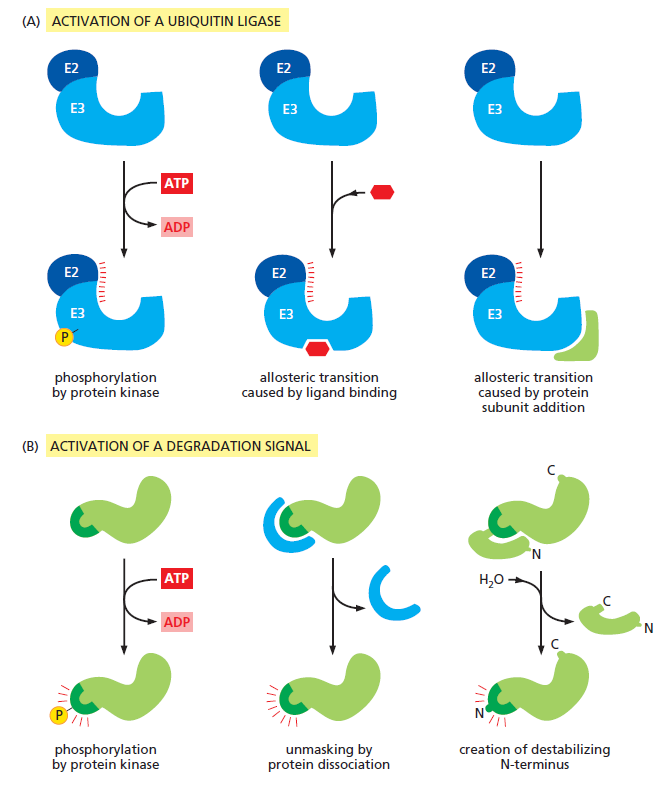
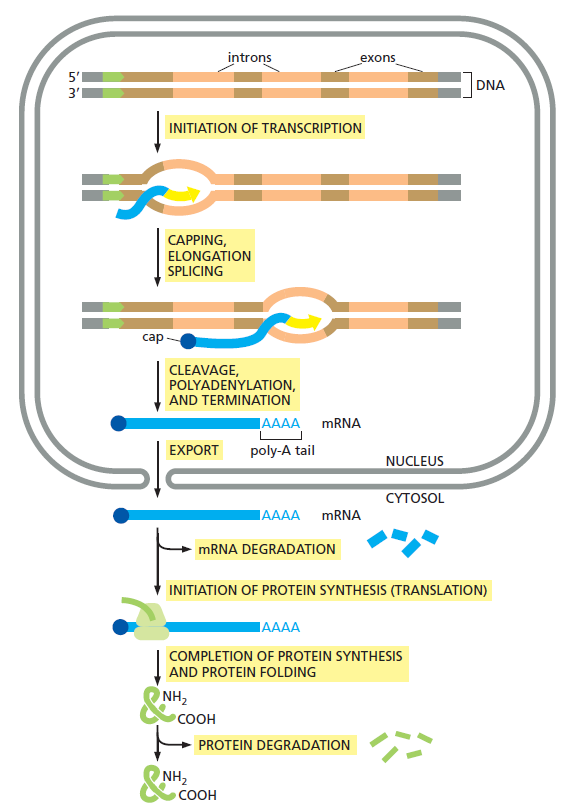
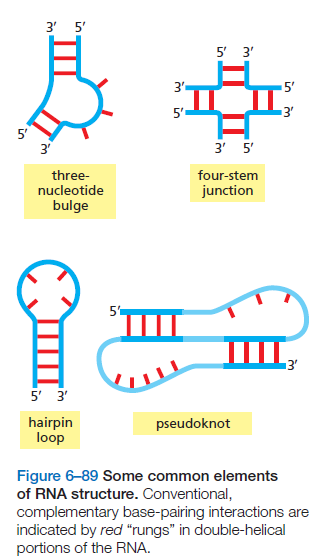
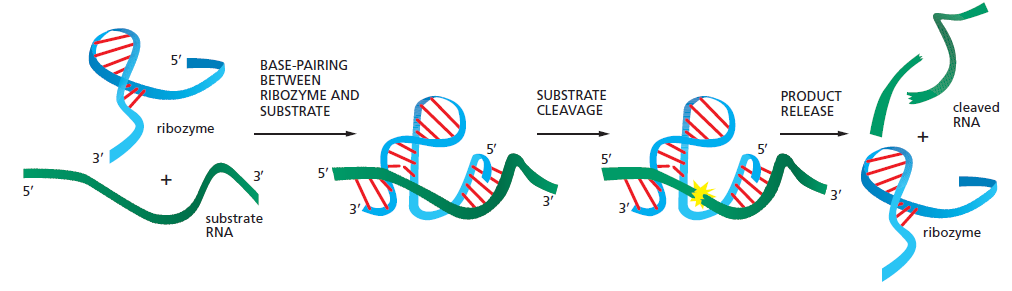
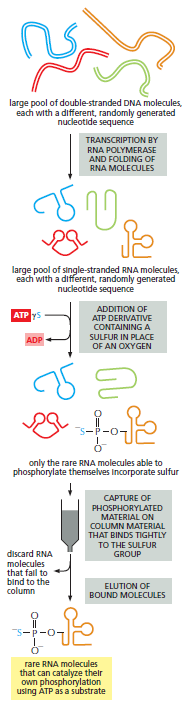
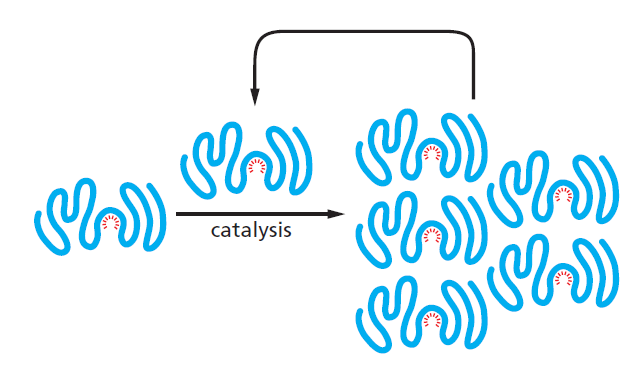
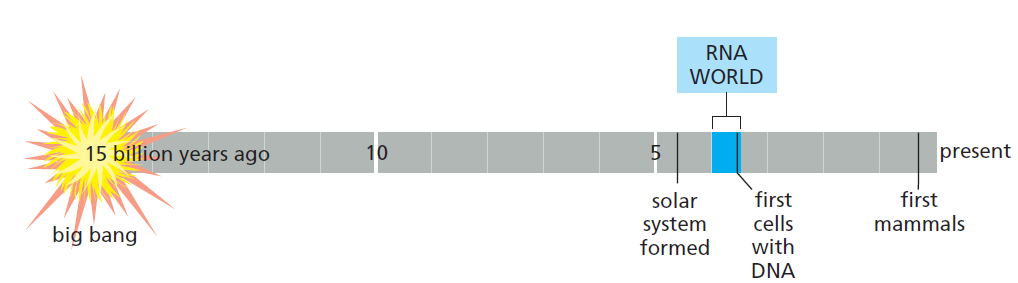
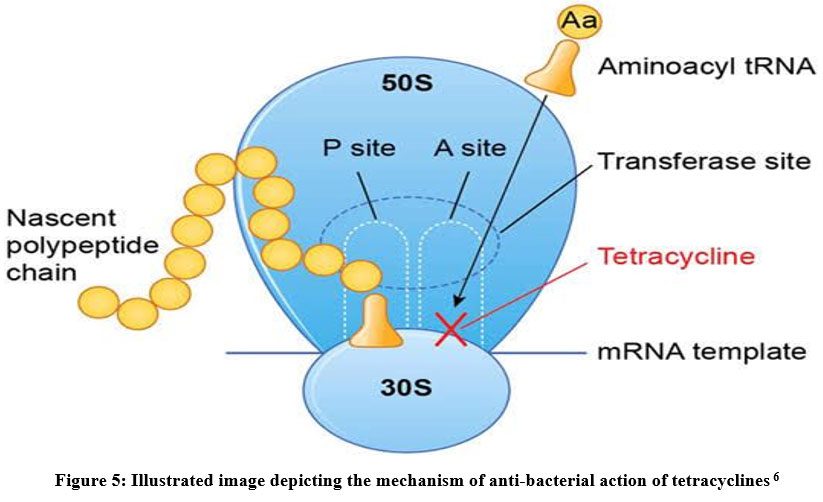
课堂问题:四环素选择性靶向细菌而非真核细胞的分子基础是什么?细菌为何能通过外排泵快速产生耐药?
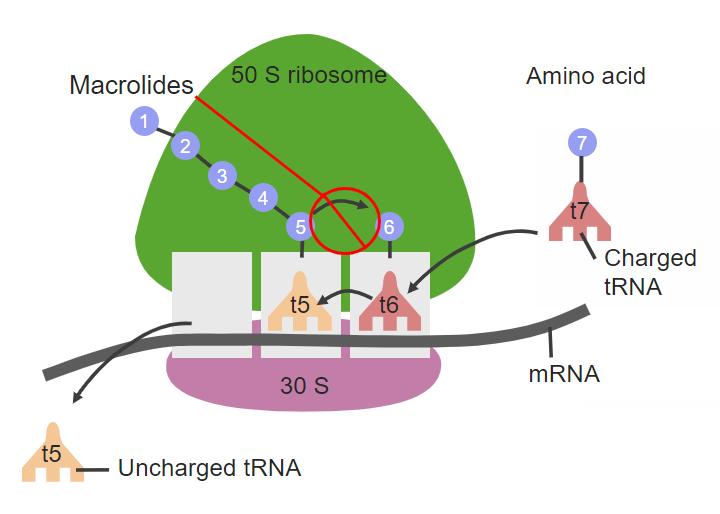
课堂问题:23S rRNA A2058位甲基化如何从结构上解释MLSB耐药表型的形成?
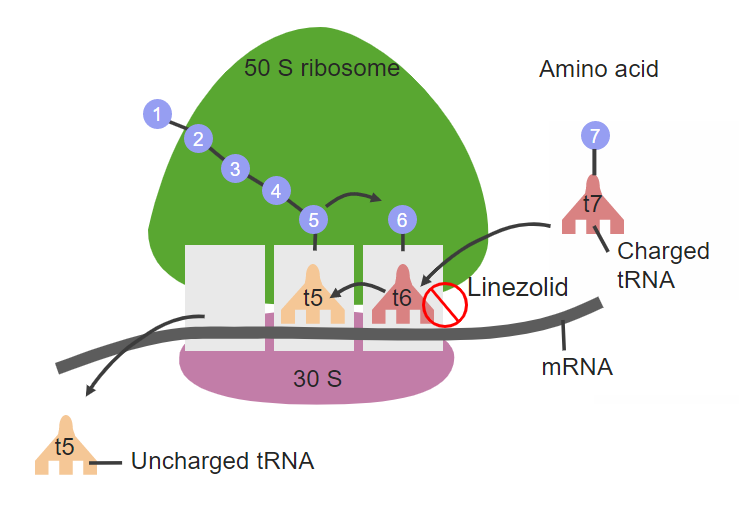
课堂问题:利奈唑胺耐药率仍相对较低,与其靶位(23S rRNA)改变需要多拷贝突变积累有何关系?
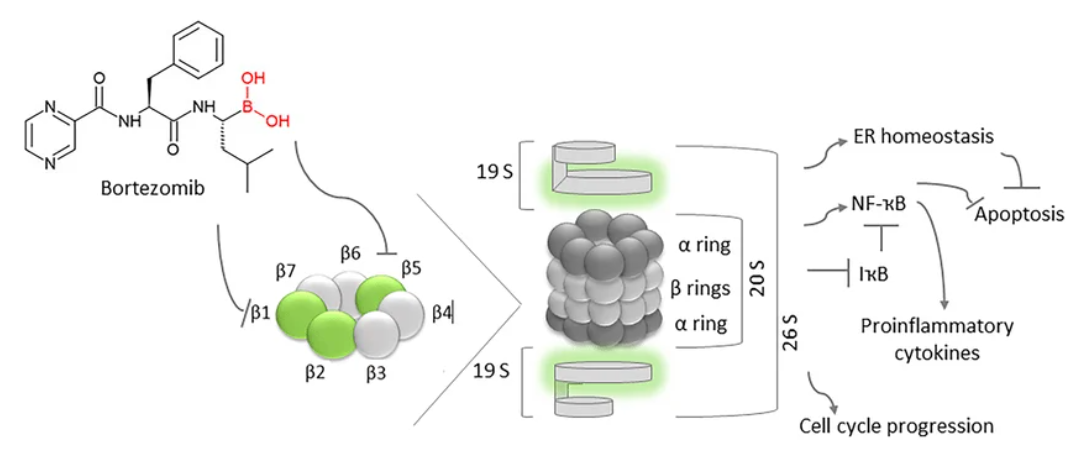
课堂问题:为什么多发性骨髓瘤对蛋白酶体抑制剂特别敏感,而大多数实体肿瘤响应有限?
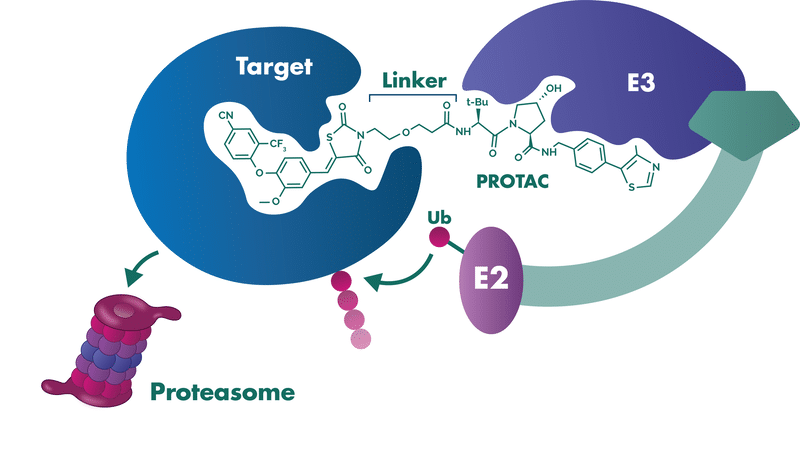

课堂问题:PROTAC 不直接抑制酶活而是诱导降解,为什么能克服传统小分子抑制剂的耐药?对于本身没有酶活的转录因子(如 MYC、AR),PROTAC 提供了什么独特机会?
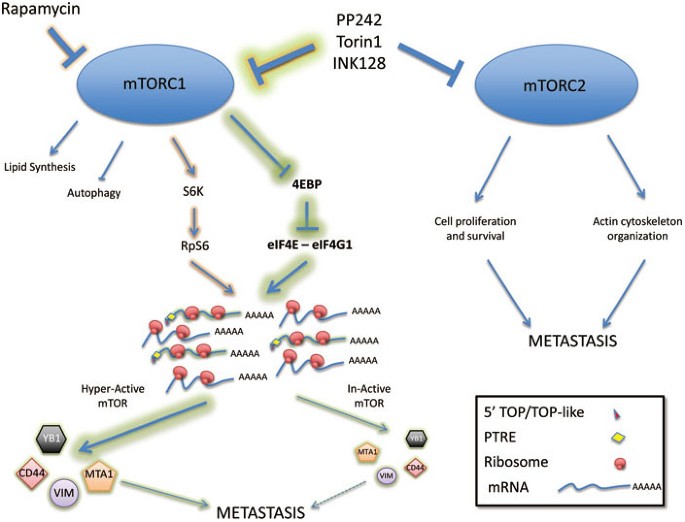
课堂问题:mTOR抑制剂对翻译的影响具有mRNA选择性,哪类mRNA的翻译最依赖eIF4E?
课堂问题:密码子优化如何提高mRNA药物的翻译效率?与原始序列相比,优化序列在哪些环节改变了翻译动力学?
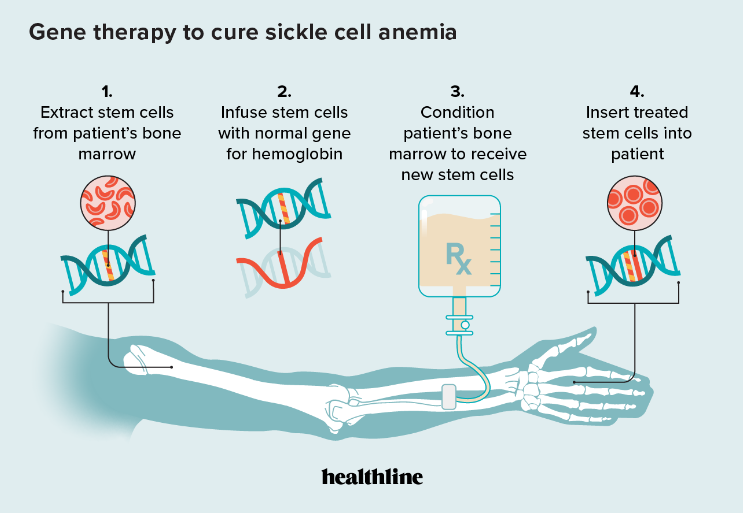
课堂问题:AAV基因疗法和mRNA疗法在持久性、免疫原性和生产复杂度上有何根本差异?各自适合哪类疾病?
课堂问题:siRNA沉默与反义ASO敲减同样靶向mRNA,二者在作用机制、递送方式和持续时间上有哪些本质区别?
课堂问题:siRNA疗法靶向肿瘤细胞的最大挑战之一是递送效率,LNP和GalNAc各擅长递送到哪类组织?
课堂问题:Eteplirsen只能帮助约13%的DMD患者,为什么外显子跳跃策略不能通用?突变位置与可跳跃外显子的关系是什么?
课堂问题:同样治疗SMA,AAV9(Zolgensma)与ASO(Nusinersen)在基因表达层次和治疗逻辑上有何根本不同?